1. 根据免疫组库TCRβ预测病人的CMV感染状态
这个项目用到了641个样本(cohort 1),包括352 例CMV阴性(CMV-)和289例CMV阳性(CMV+)
外部验证用到了120个样本(cohort 2)
该机器学习的任务为:

讨论 TCRβ 免疫组的数据特点:
- 可能出现的TCRβ的集合非常大,而单个样本只能从中检测到稀疏的少数几个;
- 一个新样本中很可能会出现训练样本集合中未出现的TCRβ克隆类型;
- 对于一个给定的TCR,它对给定抗原肽的结合亲和力会受到HLA类型的调控,因此原始的用于判别分析的特征集合还受到隐变量——HLA类型的影响;

特征选择:鉴定表型相关的TCRβs
使用Fisher精确检验(单尾检验,具体实现过程请查看文末 *2.1. Fisher检验筛选CMV阳性(CMV+)相关克隆):

Fisher检验的阈值设为:$P<1\times 10^{-4}$,FDR<0.14(该FDR的计算方法见文末 *3. FDR的计算方法,且阈值的选择问题本质上是一个模型选择问题 (model selection) ,该问题会在这部分靠后的位置进行讨论),且富集在CMV+样本中,从而得到与CMV+相关的CDR3克隆集合,共有164个
通过下面的TCRβ克隆在两组中的发生率的散点图可以明显地看到,筛出的表型相关的TCRβ克隆的确显著地表达在CMV+组中
计算表型负荷(phenotype burden)
一个样本的表型负荷(phenotype burden)被定义为:
该样本的所有unique TCRβs中与阳性表型相关的克隆的数量所占的比例
若阳性表型相关的克隆的集合记为CDR,样本i的unique克隆集合记为CDRi,则它的表型负荷为:
$$PB_i=\frac{||CDR_i \cap CDR||}{||CDR_i||}$$
其中$||·||$表示集合
·中元素的数量下图是将上面的表型负荷计算公式中的分子与分母分别作为纵轴和横轴,画成二维的散点图

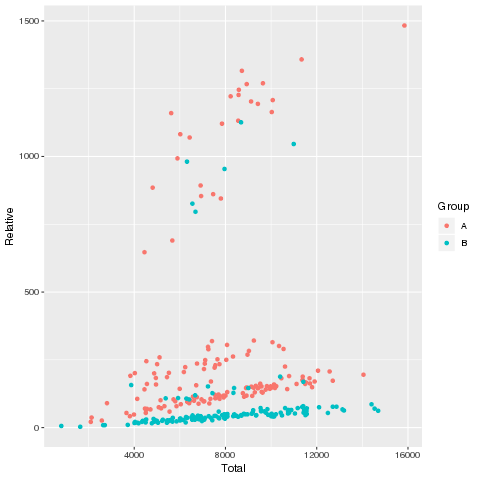
可以明显地看出两类样本在这个层面上来看,有很好的区分度
基于二项分布的贝叶斯判别模型
基本思想:
对于$CMV^+$相关TCR数为$k’$,total unique TCR数为$n’$的样本,认为它一个概率为它的表型负荷$\theta’$($\theta’$服从Beta分布),$n=n’$, $k=k’$的二项分布(伯努利分布),根据贝叶斯思想,构造最优贝叶斯分类器,即
$$h(k’,n’)=arg \max_{x \in \{+,\,-\}} p(c=x \mid k’,\,n’)$$
其中
$$p(c=x \mid k’,\,n’)=\frac{p(k’ \mid n’,\,c)p(c)}{p(k’)}$$
而$p(k’)$是一个常数,对分类器的结果没有影响,可以省略
那么就需要根据训练集估计:
- 类先验概率$p(c)$
类条件概率(似然)$p(k’ \mid n’,\,c)$
(1)首先根据概率图模型推出单个样本的概率表示公式
概率图模型如下:

则对于j样本,我们可以算出它的 $\theta_i$ 的后验分布:
$$p(\theta_i \mid y_{ij},\,\alpha_i,\,\beta_i)=\frac{p(\theta_i \mid \alpha_i,\,\beta_i)p(y_{ij} \mid \theta_i)}{p(y_{ij})}$$
其中,
- $p(y_{ij})$:表示事件$(k_{ij} \mid n_{ij})$发生的概率,即$p(k_{ij}\mid n_{ij},\,c_i)$
- $p(y_{ij} \mid \theta_i)$:表示$Binomial(k_{ij} \mid n_{ij},\,\theta_i)=\left(\begin{matrix}n_{ij}\ k_{ij}\end{matrix}\right)\theta_i^{k_{ij}}(1-\theta_i)^{n_{ij}-k_{}ij}$
- $p(\theta_i \mid \alpha_i,\,\beta_i)$:表示$\theta_i$的先验分布$Beta(\theta_i\mid\alpha_i,\,\beta_i)$
对上面的公式进一步推导
$$\begin{aligned}
&p(\theta_i \mid y_{ij},\,\alpha_i,\,\beta_i) \\
&=\frac{p(\theta_i \mid \alpha_i,\,\beta_i)p(y_{ij} \mid \theta_i)}{p(y_{ij})}\\
&=\frac{1}{p(y_{ij})}Beta(\theta_i \mid \alpha_i,\,\beta_i)Binomial(k_{ij}\mid n_{ij},\,\theta_i) \\
&=\frac{1}{p(y_{ij})} \quad {\frac{1}{B(\alpha_i,\,\beta_i)}\theta_i^{\alpha_i-1}(1-\theta_i)^{\beta_i-1}} \quad {\left(\begin{matrix}n_{ij}\\ k_{ij}\end{matrix}\right)\theta_i^{k_{ij}}(1-\theta_i)^{n_{ij}-k_{ij}}} \\
&=\frac{1}{p(y_{ij})} \quad \frac{1}{B(\alpha_i,\,\beta_i)} \left(\begin{matrix}n_{ij}\\ k_{ij}\end{matrix}\right) \theta_i^{(\alpha_i+k_{ij})-1}(1-\theta_i)^{(\beta_i+n_{ij}-k_{ij})-1} \\
&=\frac{1}{p(y_{ij})} \quad \frac{B(\alpha_i+k_{ij},\,\beta_i+n_{ij}-k_{ij})}{B(\alpha_i,\,\beta_i)} \left(\begin{matrix}n_{ij}\\ k_{ij}\end{matrix}\right) \frac{1}{B(\alpha_i+k_{ij},\,\beta_i+n_{ij}-k_{ij})} \theta_i^{(\alpha_i+k_{ij})-1}(1-\theta_i)^{(\beta_i+n_{ij}-k_{ij})-1} \\
&=\frac{1}{p(y_{ij})} \quad \frac{B(\alpha_i+k_{ij},\,\beta_i+n_{ij}-k_{ij})}{B(\alpha_i,\,\beta_i)} \left(\begin{matrix}n_{ij}\\ k_{ij}\end{matrix}\right) \quad Beta(\alpha_i+k_{ij},\,\beta_i+n_{ij}-k_{ij})
\end{aligned}
$$根据Beta分布的先验分布的,已知
$$p(\theta_i \mid y_{ij},\,\alpha_i,\,\beta_i)=Beta(\alpha_i+k_{ij},\,\beta_i+n_{ij}-k_{ij})$$
因此
$$p(k_{ij}\mid n_{ij},\,c_i)=p(y_{ij})=\left(\begin{matrix}n_{ij}\\ k_{ij}\end{matrix}\right) \frac{B(\alpha_i+k_{ij},\,\beta_i+n_{ij}-k_{ij})}{B(\alpha_i,\,\beta_i)} $$
这样我们就得到单个样本的概率表示公式,其中$B(·)$是Beta函数,从上面的表达式中,我们可以看出$p(k_{ij}\mid n_{ij},\,c_i)$是$\alpha_i$和$\beta_i$的函数
(2)优化每个组的表型负荷 $\theta_i$ 的先验分布的两个参数 $\alpha_i$ 和 $\theta_i$——最大似然法
我们要最大化$c_i$组的样本集合它们的联合概率:
$$
\begin{aligned}
p(k_i \mid n_i,\,c_i) &=\prod_{j,j \in c_i} \left(\begin{matrix}n_{ij}\\ k_{ij}\end{matrix}\right) \frac{B(\alpha_i+k_{ij},\,\beta_i+n_{ij}-k_{ij})}{B(\alpha_i,\,\beta_i)}\\
&= \prod_{j,j \in c_i} \left(\begin{matrix}n_{ij}\\ k_{ij}\end{matrix}\right) \quad \prod_{j,j \in c_i} \frac{B(\alpha_i+k_{ij},\,\beta_i+n_{ij}-k_{ij})}{B(\alpha_i,\,\beta_i)}
\end{aligned}
$$其中,$\prod_{j,\,j \in c_i} \left(\begin{matrix}n_{ij}\\ k_{ij}\end{matrix}\right)$是常数,可以省略,则
$$p(k_i \mid n_i,\,c_i)=\prod_{j,j \in c_i} \frac{B(\alpha_i+k_{ij},\,\beta_i+n_{ij}-k_{ij})}{B(\alpha_i,\,\beta_i)}$$
对它取对数,得到
$$l(\alpha_i,\,\beta_i)=\log p(k_i \mid n_i,\,c_i) \\ = \sum_{j,\,j \in c_i} \log B(\alpha_i+k_{ij}, \, \beta_i+n_{ij}-k_{ij})-N_i\log B(\alpha_i, \, \beta_i)$$
其中,$N_i$是属于$c_i$组的样本数
因此优化目标为:
$$(\alpha_i^*,\beta_i^*)=arg \, \max\limits_{\alpha_i,\,\beta_i}l(\alpha_i,\,\beta_i)$$
$l(\alpha_i,\,\beta_i)$分别对$\alpha_i$和$\beta_i$求偏导
$$\frac{\partial l}{\partial \alpha_i}=-N_i(\psi(\alpha_i)-\psi(\alpha_i+\beta_i))+\sum_{j,\,j \in c_i}(\psi(\alpha_i+k_{ij})-\psi(\alpha_i+\beta_i+n_{ij} + +k_{ij}))$$
$$\frac{\partial l}{\partial \beta_i}=-N_i(\psi(\beta_i)-\psi(\alpha_i+\beta_i))+\sum_{j,\,j \in c_i}(\psi(\beta_i+n_{ij}-k_{ij})-\psi(\alpha_i+\beta_i+n_{ij} + +k_{ij}))$$
其中,$\psi(·)$是伽马函数
使用梯度上升(gradient ascent)法来求解优化目标,其中梯度的公式为:
$$\alpha_i := \alpha_i+\alpha\frac{\partial l}{\partial \alpha_i}\\ \beta_i := \beta_i+\alpha\frac{\partial l}{\partial \beta_i}$$
最终得到的解记为$\alpha_i^*$和$\beta_i^*$,其中
$$
\alpha_+^*=4.0,\quad \beta_+^*=51,820.1\\
\alpha_-^*=26.7,\quad \beta_-^*=2,814,963.8
$$(3)根据训练好的分类器对新样本进行分类
分类器为
$$
\begin{aligned}
h(k’,n’) &= arg \max_{x \in \{CMV^+,\,CMV^-\}} p(c=x \mid n’,k’) \\
&=arg \max_{x \in \{CMV^+,\,CMV^-\}} p(k’ \mid n’, c=x)p(c=x) \\
&=arg \max_{x \in \{CMV^+,\,CMV^-\}}\left(\begin{matrix}n’ \\ k’\end{matrix}\right)\frac{B(\alpha_x^*+k’,\,\beta_x^*+n’-k’)}{B(\alpha_x^*,\,\beta_x^*)}\frac{N_x}{N}
\end{aligned}
$$(4)选择合适的的阈值:model selection
使用交叉验证法中的留一法 (leave-one-out)来进行模型选择
定义该问题的优化目标为最小化cross-entropy loss:
$$L(\phi)=-\frac{1}{N}\sum_{i=1}^N [c_i\log q_i(\phi)+(1-c_i)\log (1-q_i(\phi))]$$
其中,$c_i$表示样本$i$的所属类别,$q_i(\phi)$表示样本$i$被判为$CMV^+$的概率,N为训练样本数(N个训练样本数的训练集它总共有N种留一法组合形式)
分析优化目标为什么选择最小化cross-entropy loss:
我们的目的是选择合适的阈值,那么什么叫合适的阈值?就是在该阈值下,能筛选出合适的features,基于这些features训练出的模型能有足够高的泛化性能,即模型的variance足够的小,也就是对于测试数据(这里就是留一法中的那一个样本)有足够高的预测准确性,定量化描述就是N次留一法的准确预测的概率的均值(几何平均数)最大化,即
$$\max_{\phi} \sqrt[N]{\prod_i^N q_i(\phi)^{c_i}(1-q_i(\phi)^{1-c_i}} \\ s.t.\quad c_i \in {0,\,1}$$
由于取对数不影响优化方向,所以
$$\begin{aligned}&\log \left[\prod_i^N q_i(\phi)^{c_i}(1-q_i(\phi)^{1-c_i} \right]^{1/N} \\ &=\frac{1}{N}\log \prod_i^N q_i(\phi)^{c_i}(1-q_i(\phi)^{1-c_i} \\ &=\frac{1}{N}\sum_{i=1}^N [c_i\log q_i(\phi)+(1-c_i)\log (1-q_i(\phi))] \end{aligned}$$
从而得到上面的优化目标
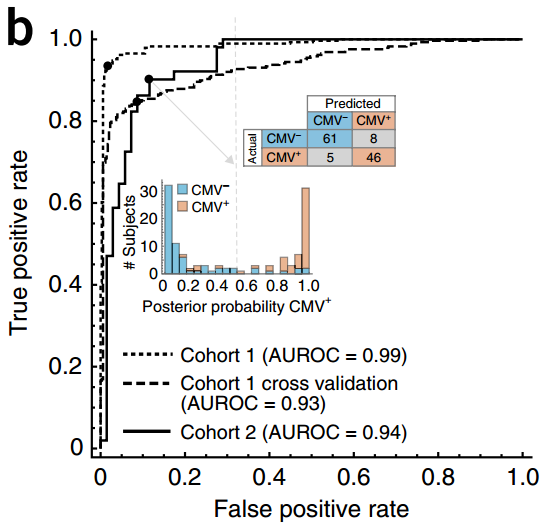
最后得到的分类器的性能非常高
2. SC3:单细胞表达谱的无监督聚类
该聚类方法名为SC3(Single-Cell Consensus Clustering)
该方法本质上就是K-means聚类,不过在执行K-means聚类的前后进行了一些特殊的操作:
- k-means聚类前:进行了数据预处理,即特征的构造,称为特征工程,该方法中是对输入的原始特征空间进行PCA变换或拉普拉斯矩阵变换,对变换后的新特征矩阵逐渐增加提取的主成分数,来构造一系列新特征;
- k-means聚类后:特征工程构造出来的一系列新特征集合,基于这些新特征集合通过k-means聚类能得到一系列不同的聚类结果,尝试对这些聚类结果总结出consensus clustering

本人比较好奇的地方是:怎么从一系列不同的聚类结果中总结出consensus clustering?
使用CSPA算法(cluster-based similarity partitioning algorithm)
(1)对每一个聚类结果按照以下方法构造二值相似度矩阵S:如果两个样本i和j在该聚类结果中被聚到同一个集合中,则它们之间的相似度为1,在二值相似度矩阵中对应的值 Si,j = 1,否则Si,j = 0;
(2)对所有的聚类结果的二值相似度矩阵S取平均,得到consensus matrix;
(3)基于consensus matrix进行层次聚类,得到最终的consensus clustering;
3. GATK的BQSR
Phred碱基质量值是由测序仪内部自带的base-calling算法评估出来的,而这种base-calling算法由于受专利保护,掌握在测序仪生成商手中,研究人员并不能了解这个算法的细节,它对于人们来说就是一个黑盒子
而测序仪的base-calling算法给出的质量评估并不十分准确,它带有一定程度的系统误差(非随机误差),使得实际测序质量值要么被低估,要么被高估
BQSR试图利用机器学习的方法来对原始的测序质量值进行校正
例如:
对于一个给定的Run,我们发现,无论什么时候我在测序一个AA 的子序列时,改子序列后紧接着的一个任意碱基的测序错误率总是要比它的实际错误率高出1%,那么我就可以将这样的碱基找出来,将它的原始测序错误率减去1%来对它进行校正
会影响测序质量评估准确性的因素有很多,主要包括序列组成、碱基在read中的位置、测序反应的cycle等等,它们以类似于叠加的形式协同产生影响,这些可能的影响因素被称作协变量 (covariable)
注意:BQSR只校正碱基质量值而不改变碱基组成,特别是对于那些质量值偏低的碱基,我们只能说它被解析成当前碱基组成的准确性很低,但是我们又无法说明它实际更可能是哪种碱基,所以干脆不改
那么,BQSR的工作原理是怎样的?
BQSR本质上是一种回归模型
前提假设:影响质量评估的因素只有reads group来源,测序的cycle和当前测序碱基的序列组成背景(这里将它上游的若干个连续位点的碱基组成看作它的背景,一般为2~6,BQSR中默认为6)
则基于这个前提假设,我们可以得出以下结论:
相同reads group来源,同处于一个cycle,且序列背景相同的碱基,它们具有相同的测序错误率,这样的碱基组成一个bin
则可以建立这样的拟合模型:
$$X_i=(RG_i,Cyc_i,Context_i) \quad \begin{matrix} f \\ \to \end{matrix} \quad y_i$$
其中,i表示当前碱基,$RG_i$表示碱基所属的Reads Group来源,$Cyc_i$表示该碱基所在的测序cycle,$Context_i$表示该碱基的序列组成背景,$y_i$表示该碱基的实际测序质量(emprical quality)
这三个分量可以直接通过输入的BAM文件的记录获得,那如何获得实际的实际测序质量呢?
可以通过BAM文件中的比对结果推出
用给定的大型基因组测序计划得到的人群变异位点作为输入,将样本中潜在变异位点与人群注释位点overlap的部分过滤掉,则剩下的那些位点,我们假设它们都是“假”的变异位点,是测序错误导致的误检
则实际测序质量为:
$$EQ=-10\log \frac{\#mismatch + 1}{\#bases + 2}$$
注意:emprical quality是以bin为单位计算出来的
这样,有了X和Y,就可以进行拟合模型的训练了,训练好的模型就可以用于碱基质量值的校正
上述只是BQSR的基本逻辑框架,在实际的实现细节上会稍有一些差别
补充知识
*1. beta分布
*1.1. 什么是beta分布
对于硬币或者骰子这样的简单实验,我们事先能很准确地掌握系统成功的概率。
然而通常情况下,系统成功的概率是未知的,但是根据频率学派的观点,我们可以通过频率来估计概率。为了测试系统的成功概率,我们做n次试验,统计成功的次数k,于是很直观地就可以计算出。然而由于系统成功的概率是未知的,这个公式计算出的只是系统成功概率的最佳估计。也就是说实际上也可能为其它的值,只是为其它的值的概率较小。因此我们并不能完全确定硬币出现正面的概率就是该值,所以也是一个随机变量,它符合Beta分布,其取值范围为0到1
用一句话来说,beta分布可以看作一个概率的概率密度分布,当你不知道一个东西的具体概率是多少时,它可以给出了所有概率出现的可能性大小。
Beta分布有和两个参数α和β,其中α为成功次数加1,β为失败次数加1。
*1.2. 理解beta分布
举一个简单的例子,熟悉棒球运动的都知道有一个指标就是棒球击球率(batting average),就是用一个运动员击中的球数除以击球的总数,我们一般认为0.266是正常水平的击球率,而如果击球率高达0.3就被认为是非常优秀的。
现在有一个棒球运动员,我们希望能够预测他在这一赛季中的棒球击球率是多少。你可能就会直接计算棒球击球率,用击中的数除以击球数,但是如果这个棒球运动员只打了一次,而且还命中了,那么他就击球率就是100%了,这显然是不合理的,因为根据棒球的历史信息,我们知道这个击球率应该是0.215到0.36之间才对啊。
对于这个问题,一个最好的方法来表示这些经验(在统计中称为先验信息)就是用beta分布,这表示在我们没有看到这个运动员打球之前,我们就有了一个大概的范围。beta分布的定义域是(0,1)这就跟概率的范围是一样的。
s
接下来我们将这些先验信息转换为beta分布的参数,我们知道一个击球率应该是平均0.27左右,而他的范围是0.21到0.35,那么根据这个信息,我们可以取α=81,β=219
之所以取这两个参数是因为:
beta分布的均值
这个分布主要落在了(0.2,0.35)间,这是从经验中得出的合理的范围

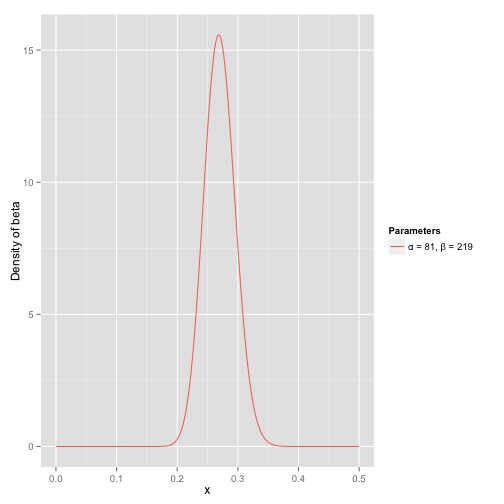
Beta(81, 219)
在这个例子里,我们的x轴就表示各个击球率的取值,x对应的y值就是这个击球率所对应的概率密度。也就是说beta分布可以看作一个概率的概率密度分布。
那么有了先验信息后,现在我们考虑一个运动员只打一次球,那么他现在的数据就是“1中;1击”。这时候我们就可以更新我们的分布了,让这个曲线做一些移动去适应我们的新信息,移动的方法很简单

其中α0和β0是一开始的参数,在这里是81和219。所以在这一例子里,α增加了1(击中了一次)。β没有增加(没有漏球)。这就是我们的新的beta分布 Beta(81+1,219),我们跟原来的比较一下:
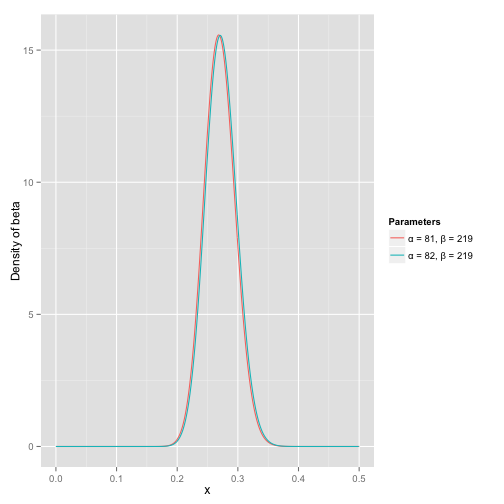
可以看到这个分布其实没多大变化,这是因为只打了1次球并不能说明什么问题。但是如果我们得到了更多的数据,假设一共打了300次,其中击中了100次,200次没击中,那么这一新分布就是 Beta(81+100,219+200) :
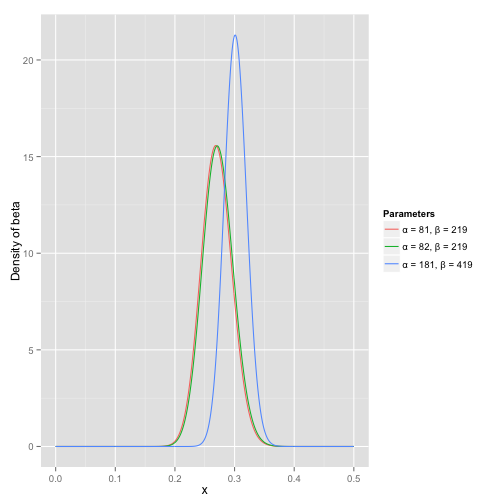
注意到这个曲线变得更加尖,并且平移到了一个右边的位置,表示比平均水平要高
有趣的现象:
根据这个新的beta分布,我们可以得出他的数学期望为:αα+β=82+10082+100+219+200=.303 ,这一结果要比直接的估计要小 100100+200=.333 。你可能已经意识到,我们事实上就是在这个运动员在击球之前可以理解为他已经成功了81次,失败了219次这样一个先验信息。
*2. 重复NG免疫组库TCRβ文章的图
*2.1. Fisher检验筛选CMV阳性(CMV+)相关克隆
目的是得到类似下面的结果:
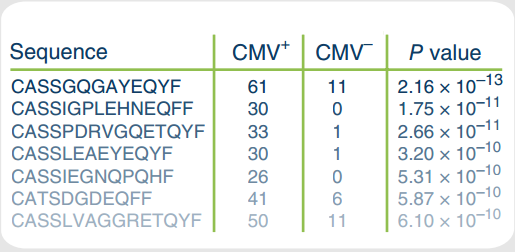
提供的输入:
- Observed matrix:行为CDR3克隆,列为样本,矩阵中的值只能取0或1,0表示该feature在该样本中未被观测到,1为被观测到了
- 样本的分组信息文件(该文件非必须,若样本名中包含分组信息,则可以不提供这个文件,只需提供分从样本名中提取分组信息的正则表达式)
- 当前感兴趣的两个组,因为考虑到可能提供的样本是多组的,而Fisher检验只能进行两组间的比较,所以多组时需要指定分析的两组是谁
实现的思路:
原始提供的输入文件是profile matrix,设定一个阈值(默认为0),将大于阈值的设为1,等于阈值的设为0,从而得到Observed matrix。这一步操作不在下面脚本的功能当中,需要自己在执行下面的脚本之前完成这个操作;
将准备好的Observed matrix输入,行为TCR克隆(feature),列为sample,因为是要对TCR克隆分析它与分组的相关性,因此每次对矩阵的行执行Fisher检验,根据提供的样本的分组信息,得到该TCR克隆的2X2列联表
$$\begin{matrix}\hline& Group \\ Observe & A & B \\ \hline Yes & a & b \\ \hline No & c & d \\ \hline \end{matrix}$$
然后再对这个列联表计算Fisher检验的p-value,将TCR克隆,列联表中的a,b和算出的p-value输出,就得到了我们想要的结果
脚本名:FisherTestForMatrix.R
1 |
|
用法:
1 | $ Rscript FisherTestForMatrix.R <profile matrix> <1|0> <group info|regx> <2 interested group> <pval> |
具体的参数说明,请查看脚本的注释信息
*2.2. 绘制表型负荷相关散点图
*2.2.1. 训练集
在上一步 *2.1. Fisher检验筛选CMV阳性(CMV+)相关克隆 的操作中会同时得到这张图
画出的表型负荷相关散点图如下:
*2.2.2. 测试集
测试集的表型负荷相关散点图需要基于训练集的结果,需要执行另外的操作
需要提供的输入:
测试集的Observed matrix:行为CDR3克隆,列为样本,矩阵中的值只能取0或1,0表示该feature在该样本中未被观测到,1为被观测到了
样本的分组信息文件(该文件非必须,若样本名中包含分组信息,则可以不提供这个文件,只需提供分从样本名中提取分组信息的正则表达式)
当前感兴趣的两个组,因为考虑到可能提供的样本是多组的,而Fisher检验只能进行两组间的比较,所以多组时需要指定分析的两组是谁
训练集的Fisher检验输出结果
脚本名:PhenotypeBurden2TestCohort.R
1 |
|
用法:
1 | $ Rscript PhenotypeBurden2TestCohort.R <profile matrix> <1|0> <group info|regx> <2 interested group> <pval> <stat> |
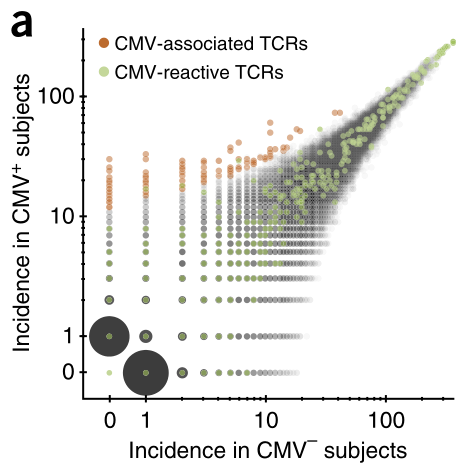
*2.3. TCRβ在两组间分布偏好性的散点图
就是画这幅图:
注意:这幅图的横纵坐标都进行了log10变换
在上一步 *2.1. Fisher检验筛选CMV阳性(CMV+)相关克隆 的操作中会同时得到这张图
画出的散点图如下:

*3. 求解NG免疫组库TCRβ文章的最优化问题
*3.1. 先验分布Beta分布的参数估计
上文中我们已经确定优化目标为
$$
(\alpha_i^*,\beta_i^*)=arg \, \max\limits_{\alpha_i,\,\beta_i}l(\alpha_i,\,\beta_i) \\
s.t. \quad \alpha,\beta \in \mathbb{R}_+
$$
其中,
$$l(\alpha_i,\,\beta_i)=\log p(k_i \mid n_i,\,c_i) \\ = \sum_{j,\,j \in c_i} \log B(\alpha_i+k_{ij}, \, \beta_i+n_{ij}-k_{ij})-N_i\log B(\alpha_i, \, \beta_i)$$
它对$\alpha$和$\beta$的偏导数分别为
$$\frac{\partial l}{\partial \alpha_i}=-N_i(\psi(\alpha_i)-\psi(\alpha_i+\beta_i))+\sum_{j,\,j \in c_i}(\psi(\alpha_i+k_{ij})-\psi(\alpha_i+\beta_i+n_{ij}+k_{ij}))$$
$$\frac{\partial l}{\partial \beta_i}=-N_i(\psi(\beta_i)-\psi(\alpha_i+\beta_i))+\sum_{j,\,j \in c_i}(\psi(\beta_i+n_{ij}-k_{ij})-\psi(\alpha_i+\beta_i+n_{ij}+k_{ij}))$$
因此可以用梯度上升法来进行求解
$$\alpha_i := \alpha_i+\alpha\frac{\partial l}{\partial \alpha_i} \\ \beta_i := \beta_i+\alpha\frac{\partial l}{\partial \beta_i}$$
先要解决一个问题:怎么用python计算Beta函数和diagama函数?
可以使用
Scipy软件包的scipy.special库来实现特殊函数的计算
special库中的特殊函数都是超越函数,所谓超越函数是指变量之间的关系不能用有限次加、减、乘、除、乘方、开方 运算表示的函数。如初等函数中的三角函数、反三角函数与对数函数、指数函数都是初等超越函数,一般来说非初等函数都是超越函数初等函数:指由基本初等函数经过有限次四则运算与复合运算所得到的函数
下面我们对
scipy.special中的部分常用函数进行说明:
$\Gamma$ (gamma)函数
$\Gamma$ 函数,也称伽玛函数,是由欧拉积分定义的函数,称为第二欧拉积分,是阶乘函数在实数域与复数域上的扩展,当 $Re(z)>0$ 时,$\Gamma$ 函数可以定义为:
$$\Gamma(z)=\int_0^{\infty}\frac{t^{z-1}}{e^t}dt$$
在
scipy.special中使用scipy.special.gamma(z)实现$\Gamma$函数的计算,如果对于精度有更高要求,可以使用采用对数坐标的scipy.special.gammaln(z)函数进行计算B函数
B函数的B是大写的希腊字母$\beta$而不是大写英文字母
B函数又称为第一类欧拉积分,第二欧拉积分则是大名鼎鼎的伽马函数,B函数的表达式和伽马函数相关
$$B(m,n)=\frac{\Gamma(m)\Gamma(n)}{\Gamma(m+n)}$$
在
scipy.special中使用scipy.special.beta(a,b)实现B函数的计算,如果对于精度有更高要求,可以使用采用对数坐标的scipy.special.betaln(a,b)函数进行计算$\psi$ (digamma)函数
Digamma函数是伽马函数的对数的导数,记为
$$\psi(x)=\frac{d \ln \Gamma(x)}{dx}=\frac{\Gamma’(x)}{\Gamma(x)}$$
在
scipy.special中使用scipy.special.digamma(z)实现digamma函数的计算Bessel(贝塞尔)函数
在介绍贝塞尔函数之前,先对贝塞尔方程进行说明,一般来说,我们将形如
$$x^2\frac{d^2y}{dx^2}+x\frac{dy}{dx}+(x^2-\alpha^2)y=0$$
的二阶线性常微分方程称为贝塞尔方程,而贝塞尔方程的标准解函数y(x)就是贝塞尔函数
其中,参数$\alpha$被称为对应贝塞尔函数的阶
*4. FDR的计算方法
*4.1. 回顾那些统计检验方法
*4.1.1. T-test与Moderated t-Test
t-test的统计量:
$$
t= \frac{\overline X_1(i)-\overline X_2(i)}{S(i)}
$$
Moderated t-Test的统计量:
$$
d= \frac{\overline X_1(i)-\overline X_2(i)}{S(i)+S_0}
$$
Moderated t-Test的统计量d与t-test的t的计算方法很相似,差别就在于分母中方差的计算方法,
|
T1 | T2 | T3 | C1 | C2 | C3 |
|---|---|---|---|---|---|---|
| Gene | XT,1 | XT,2 | XT,3 | XC,1 | XC,2 | XC,3 |
由上面展示的该基因的实际样本分组,计算出方差$S(i)=S_{X_1X_2}\sqrt{\frac{1}{n_1}+\frac{1}{n_2}}$
然后随机打乱上面的样本分组,得到:
|
T1 | T2 | T3 | C1 | C2 | C3 |
|---|---|---|---|---|---|---|
| Gene | XC,2 | XT,1 | XC,3 | XT,3 | XT,2 | XC,1 |
根据打乱的结果算出$S_0$,进行n次这样的随机打乱,计算得到$d_1,d_2,…,d_n$
最后算出它的P值:
$$P=\frac{\# \{d_i \ge d,i=1,2,…,n\}}{n} $$
之所以不用t检验的统计量查表法,是因为Moderated t-Test的统计量已经不再符合某种统计分布了,而且这样算出来的P值也具有一定的统计意义
*4.2. 多重假设检验的必要性
统计学中的假设检验的基本思路是:
设立零假设(null hypothesis)$H_0$,以及与零假设$H_0$相对应的非零假设(alternative hypothesis, or reject null hypothesis)$H_1$,在假设$H_0$成立的前提下,计算出$H_0$发生的概率,若$H_0$的发生概率很低,基于小概率事件几乎不可能发生,所以可以拒绝零假设
但是这些传统的假设检验方法研究的对象,都是一次试验
在一次试验中(注意:是一次试验, 即single test),0.05 或0.01的cutoff足够严格了(想象一下,一个口袋有100个球,95个白的,5个红的, 只让你摸一次,你能摸到红的可能性是多大?)
但是对于多次试验,又称多重假设检验,再使用p值是不恰当的,下面来分析一下为什么:
大家都知道墨菲定律:如果事情有变坏的可能,不管这种可能性有多小,它总会发生
用统计的语言去描述墨菲定律:
在数理统计中,有一条重要的统计规律:假设某意外事件在一次实验(活动)中发生的概率为p(p>0),则在n次实验(活动)中至少有一次发生的概率为 $p_n=1-(1-p)^n$
由此可见,无论概率p多么小(即小概率事件),当n越来越大时,$p_n$越来越接近1
这和我们的一句俗语非常吻合:常在河边走,哪有不湿鞋;夜路走多了,总能碰见鬼
在多重假设检验中,我们一般关注的不再是每一次假设检验的准确性,而是控制在作出的多个统计推断中犯错误的概率,即False Discover Rate(FDR),这对于医院的诊断情景下尤其重要:
假如有一种诊断艾滋病(AIDS)的试剂,试验验证其准确性为99%(每100次诊断就有一次false positive)。对于一个被检测的人(single test))来说,这种准确性够了;但对于医院 (multiple test))来说,这种准确性远远不够
因为每诊断10 000个个体,就会有100个人被误诊为艾滋病(AIDS),每一个误诊就意味着一个潜在的医疗事故和医疗纠纷,对于一些大型医院,一两个月的某一项诊断的接诊数就能达到这个级别,如果按照这个误诊率,医院恐怕得关门,所以医院需要严格控制误诊的数量,宁可错杀一万也不能放过一个,因为把一个没病的病人误判为有病,总比把一个有病的病人误判为没病更保险一些
100 independent genes. (We have 100 hypotheses to test)
No significant differences in gene expression between 2 classes (H0 is true). Thus, the probability that a particular test (say, for gene 1) is declared significant at level 0.05 is exactly 0.05. (Probability of reject H0 in one test if H0 is true = 0.05)
However, the probability of declaring at least one of the 100 hypotheses false (i.e. rejecting at least one, or finding at least one result significant) is:
$$1-(1-0.05)^{100}\approx 0.994$$
*4.3. 如何计算FDR?
统计检验的混淆矩阵:
|
$H_0$ is true | $H_1$ is true | Total |
|---|---|---|---|
| Significant | V | S | R |
| Not Significant | U | T | m-R |
| Total | m0 | m-m0 | m |
FWER (Family Wise Error Rate)
作出一个或多个假阳性判断的概率
$$FWER=Pr(V\ge 1)$$
使用这种方法的统计学过程:
- The Bonferroni procedure
- Tukey’s procedure
- Holm’s step-down procedure
FDR (False Discovery Rate)
在所有的单检验中作出假阳性判断比例的期望
$$FDR=E\left[\frac{V}{R}\right]$$
使用这种方法的统计学过程:
- Benjamini–Hochberg procedure
- Benjamini–Hochberg–Yekutieli procedure
*4.3.1. Benjamini-Hochberg procedure (BH)
对于m个独立的假设检验,它们的P-value分别为:$p_i,i=1,2,…,m$
(1)按照升序的方法对这些P-value进行排序,得到:
$$p_{(1)} \le p_{(2)} \le … \le p_{(m)}$$
(2)对于给定是统计显著性值$\alpha \in (0,1)$,找到最大的k,使得
$$p_{(k)} \le \frac{\alpha * k}{m}$$
(3)对于排序靠前的k个假设检验,认为它们是真阳性 (positive )
即:$reject \, H_0^{(i)},\, 1 \le i \le k$
$$
\begin{array}{c|l}
\hline
Gene & p-value \\
\hline
G1 & P1 =0.053 \\
\hline
G2 & P2 =0.001 \\
\hline
G3 & P3 =0.045 \\
\hline
G4 & P4 =0.03 \\
\hline
G5 & P5 =0.02 \\
\hline
G6 & P6 =0.01 \\
\hline
\end{array}
\, \Rightarrow \,
\begin{array}{c|l}
\hline
Gene & p-value \\
\hline
G2 & P(1) =0.001 \\
\hline
G6 & P(2) =0.01 \\
\hline
G5 & P(3) =0.02 \\
\hline
G4 & P(4) =0.03 \\
\hline
G3 & P(5) =0.045 \\
\hline
G1 & P(6) =0.053 \\
\hline
\end{array}
$$
$$\alpha = 0.05$$
$P(4) =0.03<0.05*\frac46=0.033$
$P(5) =0.045>0.05*\frac56=0.041$
因此最大的k为4,此时可以得出:在FDR<0.05的情况下,G2,G6,G5 和 G4 存在差异表达
可以计算出q-value:
$$p_{(k)} \le \frac{\alpha*k}{m} \, \Rightarrow \, \frac{p_{(k)}*m}{k} \le \alpha$$
| Gene | P | q-value |
|---|---|---|
| G2 | P(1) =0.001 | 0.006 |
| G6 | P(2) =0.01 | 0.03 |
| G5 | P(3) =0.02 | 0.04 |
| G4 | P(4) =0.03 | 0.045 |
| G3 | P(5) =0.045 | 0.054 |
| G1 | P(6) =0.053 | 0.053 |
根据q-valuea的计算公式,我们可以很明显地看出:
$$q^{(i)}=p_{(k)}^{(i)}*\frac{Total \, Gene \, Number}{rank(p^{(i)})}=p_{(k)}^{(i)}*\frac{m}{k}$$
即,根据该基因p值的排序对它进行放大,越靠前放大的比例越大,越靠后放大的比例越小,排序最靠后的基因的p值不放大,等于它本身
我们也可以从可视化的角度来看待这个问题:
对于给定的$\alpha \in (0,1)$,设函数$y=\frac{\alpha}{m}x \quad (x=1,2,…,m)$,画出这条线,另外对于每个基因,它在图上的坐标为$(rank(p_{(k)}^{(i)}),p_{(k)}^{(i)})=(k,p_{(k)}^{(i)})$,图如下:
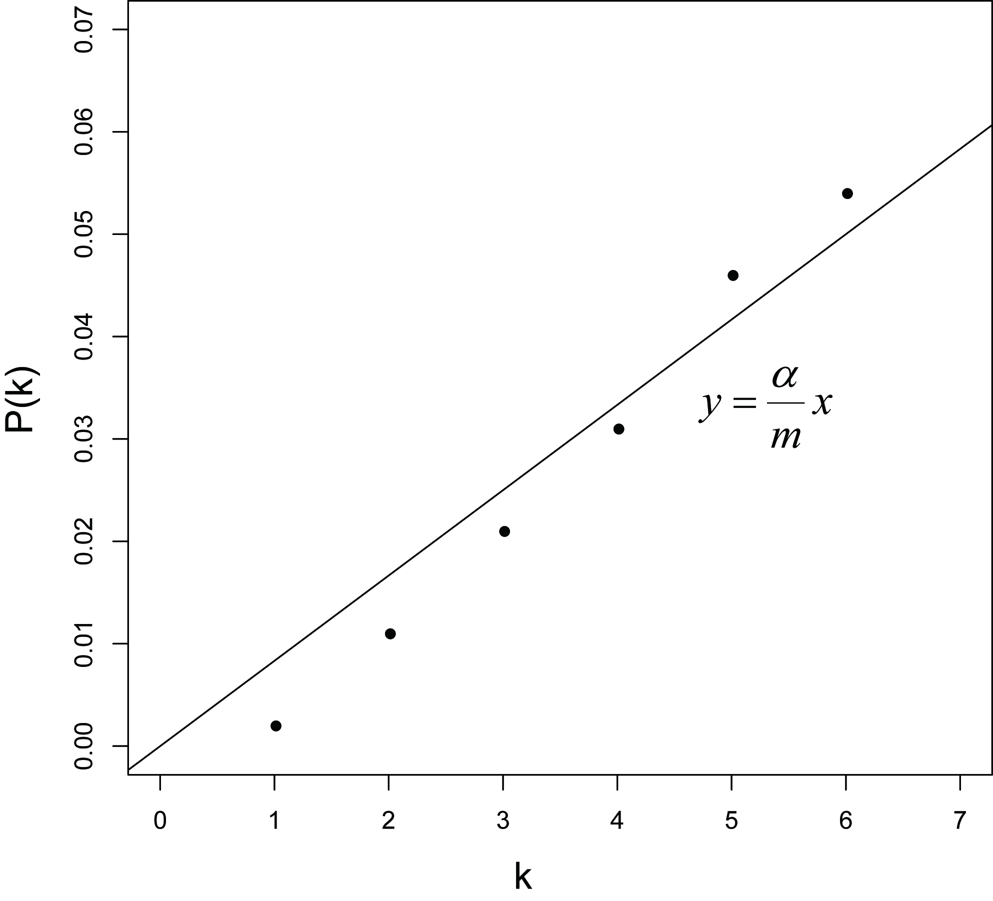
通过设置$\alpha$可以改变图中直线的斜率,$\alpha$越大,则直线的斜率越大,落在直线下方的点就越多,通过FDR检验的基因也就越多,反之,直线的斜率越小,落在直线下方的点就越少,通过FDR检验的基因也就越少
*4.4.2. Bonferroni 校正
Bonferroni 校正的基本思想:
如果在同一数据集上同时检验n个独立的假设,那么用于每一假设的统计显著水平,应为仅检验一个假设时的显著水平的1/n
举个例子:如要在同一数据集上检验两个独立的假设,显著水平设为常见的0.05,此时用于检验该两个假设应使用更严格的0.025,即0.05* (1/2)
序列化的 Bonferroni 校正步骤:
对k个独立的检验,在给定的显著性水平α下,把每个检验对应的 P 值从小到大排列
$$p_{(1)} \le p_{(2)} \le … \le p_{(k)}$$
首先看最小的 P 值 $p_{(1)}$,如果$p_{(1)} \le \frac{\alpha}{k}$,就认为对应的检验在总体上(table wide)α水平上显著;如果不是,就认为所有的检验都不显著;
当且仅当 $p_{(1)} \le \frac{\alpha}{k}$ 时,再来看第二个P值$p_{(2)}$。如果$p_{(2)} \le \frac{\alpha}{k-1}$,就认为在总体水平上对应的检验在α水平上是显著的;
之后再进行下一个P值……一直进行这个过程,直到 $p_{(i)} \le \frac{\alpha}{k-i+1}$不成立;下结论i和以后的检验都不显著
*5. 如何在Python中实现几种主要的概率分布
用 scipy模块 可以实现这些概率分布,具体实现方法请参考文章 如何在Python中实现这五类强大的概率分布
常见的概率分布包括离散概率分布和连续概率分布
离散概率分布也称为概率质量函数(probability mass function, pmf)
连续概率分布也称为概率密度函数(probability density function, pdf)
离散分布有:
- 二项分布 (Binomial Distribution)
- 泊松分布 (Poisson Distribution)
连续分布有:
- 正态分布 (Normal Distribution)
- B分布 (Beta Distribution)
- 指数分布(Exponential Distribution)
二项分布
$$P(X=k)=\left(\begin{matrix}n \ k\end{matrix}\right) p^k(1-p)^{n-k}$$
$$E(X)=np,\quad Var(X)=np(1-p)$$
1
2
3
4
5
6
7
8
9
10n = 10
p = 0.3
k = np.arange(0, 21)
binomial = stats.binom.pmf(k, n, p)
plt.plt(k, binomial, 'o-')
plt.title('Binomial: n=%i p=%.2f'%(n, p), fontsize=20)
plt.xlabel('Number of success', fontsize=15)
plt.ylabel('Probability of success', fontsize=15)
plt.show()
可以使用
.rvs函数模拟一个二项随机变量,其中参数size指定你要进行模拟的次数。让Python返回10000个参数为n和p的二项式随机变量。输出这些随机变量的平均值和标准差,然后画出所有的随机变量的直方图1
2
3
4
5
6
7binom_sim = stats.binom.rvs(n=10, p=0.3, size=10000)
print('Mean: %f'%np.mean(binom_sim))
print('SD: %f'%np.std(binom_sim))
plt.hist(binom_sim, bins=10, density=True)
plt.xlabel('X')
plt.ylabel('density')
plt.show()
由于绘图部分的代码基本相同,所以下面部分只展示数据生成的代码
泊松分布
$$P(X=k)=\frac{\lambda^ke^{-\lambda}}{k!}$$
$$E(X)=\lambda,\quad Var(X)=\lambda$$
考虑平均每天发生2起事故的例子:
1
2
3
4
5
6
7
8rate = 2
n = np.arange(0, 10)
y = stats.poisson.pmf(n, rate)
# 模拟泊松分布
data = stats.poisson.rvs(mu=2, loc=0, size=1000)
print('Mean: %f'%np.mean(data))
print('SD: %f'%np.std(data, ddof=1))
正态分布 (Normal Distribution)
$$P(x \mid \mu,\sigma)=\frac{1}{\sqrt{2\pi}\sigma}\exp\left(-\frac{(x-\mu)^2}{2\sigma^2}\right)$$
$$E(X)=\mu,\quad Var(X)=\sigma^2$$
1
2
3
4mu = 0
sigma = 1
x = np.arange(-5, 5, 0.1)
y = stats.norm.pdf(x, 0, 1)
$\beta$分布
β分布是一个取值在 [0, 1] 之间的连续分布,它由两个形态参数 α 和 β 的取值所刻画
$$
\begin{aligned}
&P(x\mid \alpha,\beta) \\
&=\frac{1}{B(\alpha,\beta)}x^{\alpha-1}(1-x)^{\beta-1} \\
&=\frac{\Gamma(\alpha+\beta)}{\Gamma(\alpha)\Gamma(\beta)}x^{\alpha-1}(1-x)^{\beta-1} \quad x\in[0,1],\,\alpha>0,\beta>0
\end{aligned}
$$$$E(X)=\frac{\alpha}{\alpha+\beta},\quad Var(X)=\frac{\alpha\beta}{(\alpha+\beta)^2(\alpha+\beta+1)}$$
贝叶斯分析中大量使用了$\beta$分布
1
2
3
4a = 0.5
b = 0.5
x = np.arange(0.01, 1, 0.01)
y = stats.beta.pdf(x, a, b)
当将参数都设为1时,该分布为均匀分布
指数分布 (Exponential Distribution)
指数分布是一种连续概率分布,用于表示独立随机事件发生的时间间隔。比如旅客进入机场的时间间隔、打进客服中心电话的时间间隔、中文维基百科新条目出现的时间间隔等等
$$P(x\mid \lambda)=\lambda e^{-\lambda x} \quad x\in[0,\infty),\,\lambda>0$$
$$E(X)=1/\lambda,\quad Var(X)=1/\lambda^2$$
1
2
3
4
5
6
7
8
9lambda = 0.5
x = np.arange(0, 15, 0.1)
y = stats.expon.pdf(x, lambda)
# y = lambda * exp(-lambda * x)
# 模拟
data = stats.expon.rvs(scale=2, size=1000) # scale表示λ的倒数
print('Mean: %f'%np.mean(data))
print('SD: %f'%np.std(data, ddof=1))
参考资料:
(1) Emerson R O , Dewitt W S , Vignali M , et al. Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T cell repertoire[J]. Nature Genetics, 2017, 49(5):659-665.
(2) Kiselev, V. Y. et al. SC3: consensus clustering of single-cell RNA-seq data[J]. Nat. Methods 14, 483–486 (2017).
(3) GATK官网BQSR算法说明
(4) GATK官网VQSR算法说明
(5) Alipanahi B , Delong A , Weirauch M T , et al. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning[J]. Nature Biotechnology, 2015.
(6) CSDN·chivalry《二项分布和Beta分布》
(8) 贝塔分布(Beta Distribution)简介及其应用
(9) StatLect《Beta distribution》
(10) 简书·殉道者之花火《Python机器学习及分析工具:Scipy篇》
(11) Scipy官方文档
(13) Storey, J.D. & Tibshirani, R. Statistical signifcance for genomewide studies.Proc. Natl. Acad. Sci. USA 100, 9440–9445 (2003)
(14) 国科大研究生课程《生物信息学》,陈小伟《基因表达分析》
(16) 简书·Honglei_Ren《多重检验中的FDR错误控制方法与p-value的校正及Bonferroni校正》
(18) SELEX-Illumina