GATK4流程
![]()
准备配套数据
要明确你的参考基因组版本了!!! b36/b37/hg18/hg19/hg38,记住b37和hg19并不是完全一样的,有些微区别哦!!!
1、下载hg19
这个下载地址非常多,常用的就是NCBI,ensembl和UCSC了,但是这里推荐用这个脚本下载(下载源为UCSC):
1 | # 一个个地下载hg19的染色体 |
BWA: Map to Reference
建立参考序列索引
1
$ bwa index -a bwtsw ref.fa
参数
-a用于指定建立索引的算法:- bwtsw 适用于>10M
- is 适用于参考序列<2G (默认-a is)
可以不指定
-a参数,bwa index会根据基因组大小来自动选择合适的索引方法序列比对
1
2$ bwa mem ref.fa sample_1.fq sample_2.fq -R '@RG\tID:sample\tLB:sample\tSM:sample\tPL:ILLUMINA' \
2>sample_map.log | samtools sort -@ 20 -O bam -o sample.sorted.bam 1>sample_sort.log 2>&1-R选项为必须选项,用于定义头文件中的SAM/BAM文件中的read group和sample信息1
2
3
4
5
6
7> The file must have a proper bam header with read groups. Each read group must contain the platform (PL) and sample (SM) tags.
> For the platform value, we currently support 454, LS454, Illumina, Solid, ABI_Solid, and CG (all case-insensitive)
>
> The GATK requires several read group fields to be present in input files and will fail with errors if this requirement is not satisfied
>
> ID:输入reads集的ID号; LB: reads集的文库名; SM:样本名称; PL:测序平台
>查看SAM/BAM文件中的read group和sample信息:
1
2
3
4
5
6
7$ samtools view -H /path/to/my.bam | grep '^@RG'
@RG ID:0 PL:solid PU:Solid0044_20080829_1_Pilot1_Ceph_12414_B_lib_1_2Kb_MP_Pilot1_Ceph_12414_B_lib_1_2Kb_MP LB:Lib1 PI:2750 DT:2008-08-28T20:00:00-0400 SM:NA12414 CN:bcm
@RG ID:1 PL:solid PU:0083_BCM_20080719_1_Pilot1_Ceph_12414_B_lib_1_2Kb_MP_Pilot1_Ceph_12414_B_lib_1_2Kb_MP LB:Lib1 PI:2750 DT:2008-07-18T20:00:00-0400 SM:NA12414 CN:bcm
@RG ID:2 PL:LS454 PU:R_2008_10_02_06_06_12_FLX01080312_retry LB:HL#01_NA11881 PI:0 SM:NA11881 CN:454MSC
@RG ID:3 PL:LS454 PU:R_2008_10_02_06_07_08_rig19_retry LB:HL#01_NA11881 PI:0 SM:NA11881 CN:454MSC
@RG ID:4 PL:LS454 PU:R_2008_10_02_17_50_32_FLX03080339_retry LB:HL#01_NA11881 PI:0 SM:NA11881 CN:454MSC
...read group信息不仅会加在头信息部分,也会在比对结果的每条记录里添加一个
RG:Z:*标签1
2
3
4
5
6$ samtools view /path/to/my.bam | grep '^@RG'
EAS139_44:2:61:681:18781 35 1 1 0 51M = 9 59 TAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAA B<>;==?=?<==?=?=>>?>><=<?=?8<=?>?<:=?>?<==?=>:;<?:= RG:Z:4 MF:i:18 Aq:i:0 NM:i:0 UQ:i:0 H0:i:85 H1:i:31
EAS139_44:7:84:1300:7601 35 1 1 0 51M = 12 62 TAACCCTAAGCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAA G<>;==?=?&=>?=?<==?>?<>>?=?<==?>?<==?>?1==@>?;<=><; RG:Z:3 MF:i:18 Aq:i:0 NM:i:1 UQ:i:5 H0:i:0 H1:i:85
EAS139_44:8:59:118:13881 35 1 1 0 51M = 2 52 TAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAA @<>;<=?=?==>?>?<==?=><=>?-?;=>?:><==?7?;<>?5?<<=>:; RG:Z:1 MF:i:18 Aq:i:0 NM:i:0 UQ:i:0 H0:i:85 H1:i:31
EAS139_46:3:75:1326:2391 35 1 1 0 51M = 12 62 TAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAA @<>==>?>@???B>A>?>A?A>??A?@>?@A?@;??A>@7>?>>@:>=@;@ RG:Z:0 MF:i:18 Aq:i:0 NM:i:0 UQ:i:0 H0:i:85 H1:i:31
...若原始SAM/BAM文件没有read group和sample信息,可以通过AddOrReplaceReadGroups添加这部分信息
1
2
3
4
5
6
7
8$ java -jar picard.jar AddOrReplaceReadGroups \
I=input.bam \
O=output.bam \
RGID=4 \
RGLB=lib1 \
RGPL=illumina \
RGPU=unit1 \
RGSM=20
前期处理
在进行本部分的操作之前先要做好以下两部的准备工作
1、创建GATK索引。用Samtools为参考序列创建一个索引,这是为方便GATK能够快速地获取fasta上的任何序列做准备
1 | $ samtools faidx database/chr17.fa # 该命令会在chr17.fa所在目录下创建一个chr17.fai索引文件 |
2、生成.dict文件
1 | $ gatk CreateSequenceDictionary -R database/chr17.fa -O database/chr17.dict |
GATK4的调用语法:
1 | gatk [--java-options "-Xmx4G"] ToolName [GATK args] |
去除PCR重复
duplicates的产生原因
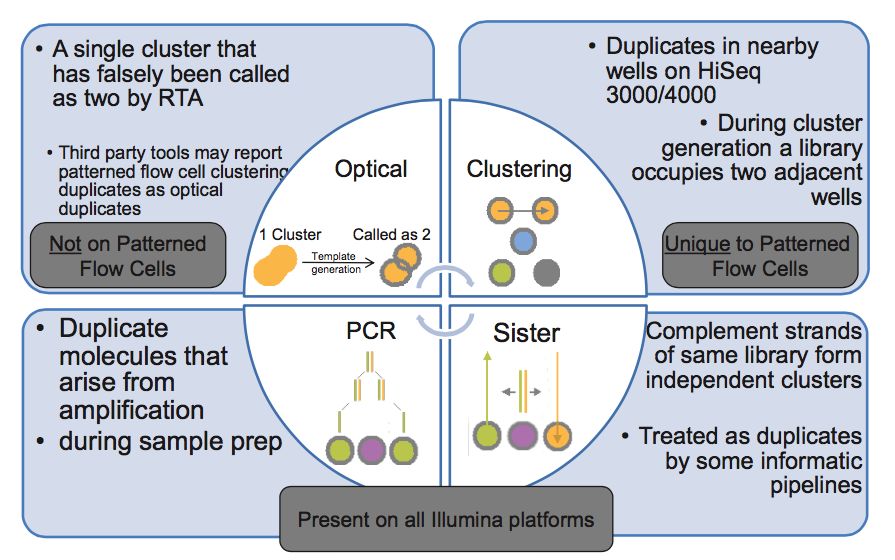
- PCR duplicates(PCR重复)
PCR扩增时,同一个DNA片段会产生多个相同的拷贝,第4步测序的时候,这些来源于同!一!个!拷贝的DNA片段会结合到Fellowcell的不同位置上,生成完全相同的测序cluster,然后被测序出来,这些相同的序列就是duplicate
- Cluster duplicates
生成测序cluster的时候,某一个cluster中的DNA序列可能搭到旁边的另一个cluster的生成位点上,又再重新长成一个相同的cluster,这也是序列duplicate的另一个来源,这个现象在Illumina HiSeq4000之后的Flowcell中会有这类Cluster duplicates
- Optical duplicates(光学重复)
某些cluster在测序的时候,捕获的荧光亮点由于光波的衍射,导致形状出现重影(如同近视散光一样),导致它可能会被当成两个荧光点来处理。这也会被读出为两条完全相同的reads
- Sister duplicates
它是文库分子的两条互补链同时都与Flowcell上的引物结合分别形成了各自的cluster被测序,最后产生的这对reads是完全反向互补的。比对到参考基因组时,也分别在正负链的相同位置上,在有些分析中也会被认为是一种duplicates。
用泊松分布解释 NGS 测序数据的 duplication 问题
我曾经有这样的疑惑,为什么文库构建过程中的 PCR 将每个文库分子都扩增了上千倍,以 PCR 10个循环为例 210= 1024 ,但是实际测序数据中 duplication 率并不高(低于20%)
有一篇文章从统计概率的角度详细探讨了一下 duplication 率的影响因素
PCR 的过程中不同长度的文库分子被扩增的效率不同(GC 太高或 AT 含量太高都会影响扩增效率),PCR 更倾向于扩增短片段的文库分子,这里先不考虑文库片段扩增效率的差异,把问题简化一下,假设所有文库分子扩增效率都相同。PCR duplicate 的主要来源是同一个文库分子的不同拷贝都在 flowcell 上生成了可以被测序的 cluster ,导致同一个分子的序列被测序仪读取多次。那么为何在每个分子都有上千个拷贝的情况下,实际却很少出现同一分子的多个拷贝被测序的情况呢?主要原因就是文库中 unique 分子的数量比被 flowcell 上引物捕获的分子数量多很多,直白点说就是 flowcell 上用于捕获文库分子的引物数量太少了,两者不在同一个数量级,导致很少出现同一个文库分子的多个拷贝被 flowcell 上引物捕获生成 cluster。
假设文库中所有分子与引物的结合都是随机的,简化一下就相当于,一个箱子中有 n 种颜色的球(文库中的 n 种 unique 分子),每种颜色有 1000 个(PCR 扩增的,随 cycle 数变化),从这个箱子中随机拿出来 k 个球(最终测序得到 k 条 reads),其中出现相同颜色的球就是 duplicate,那么 duplication 率就可以根据有多少种颜色的球被取出 0,1,2,3…… 次的概率计算,可以近似用泊松分布模型来描述。
以人全基因组重测序 30X 为例,PE150 需要约 3x108条 reads ,文库中 unique 分子数其实可以通过上机文库的浓度和体积(外加 PCR 循环数)计算出来,这里用近似值 3.5x1010 个 unique 分子。每个 unique 分子期望被测序的次数是 3x108/3.5x1010 = 0.0085 ,每个 unique 分子被测 0,1,2,3… 次的概率如下图:
1 | > x <- seq(0,10,1) |
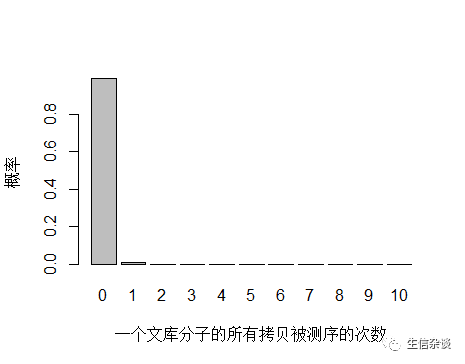
由于 unique 分子数量太多,被测 0 次的概率远高于 1 和 2 次,我们去除 0 次的看一下:
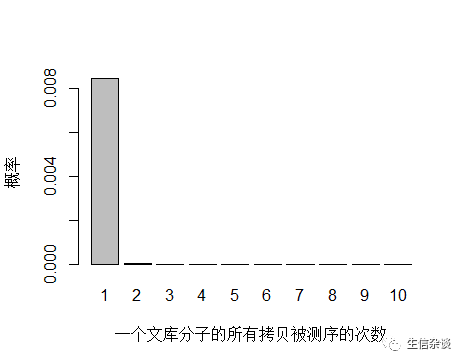
unique 分子被测序 1 次的概率远大于 2次及以上,即便一个 unique 分子被测序 2 次,我们去除 duplicate 时候还会保留其中一条 reads。
如果降低文库中 unique 分子数量到 4.5x109个,PCR 循环数增加以便浓度达到跟上面模拟的情况相同,测序 reads 数还是 3x108 条,每个 unique 分子预期被测序的次数是 3x108/4.5x109 = 0.067 。
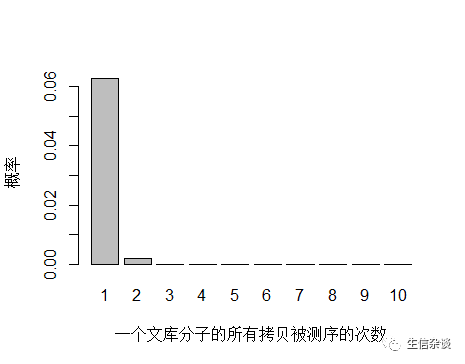
unique 分子数量减少,被测序 2次的概率增大,duplication 率显然也会增高。
到这里已经可以很明白的看出 duplication 率主要与文库中 unique 分子数量有关,所以建库过程中最大化 unique 分子数是降低 duplication 率的关键。文库中 unique 分子数越多,说明建库起始量越高,需要 PCR 的循环数越少,而文库中 unique 分子数越少,说明建库起始量越低,需要 PCR 的循环数越多,因此提高建库起始量是关键。
PCR bias的影响
DNA在打断的那一步会发生一些损失, 主要表现是会引发一些碱基发生颠换变换(嘌呤-变嘧啶或者嘧啶变嘌呤) , 带来假的变异。 PCR过程会扩大这个信号, 导致最后的检测结果中混入了假的结果;
PCR反应过程中也会带来新的碱基错误。 发生在前几轮的PCR扩增发生的错误会在后续的PCR过程中扩大, 同样带来假的变异;
对于真实的变异, PCR反应可能会对包含某一个碱基的DNA模版扩增更加剧烈(这个现象称为PCR Bias) 。 因此, 如果反应体系是对含有reference allele的模板扩增偏向强烈, 那么变异碱基的信息会变小, 从而会导致假阴。
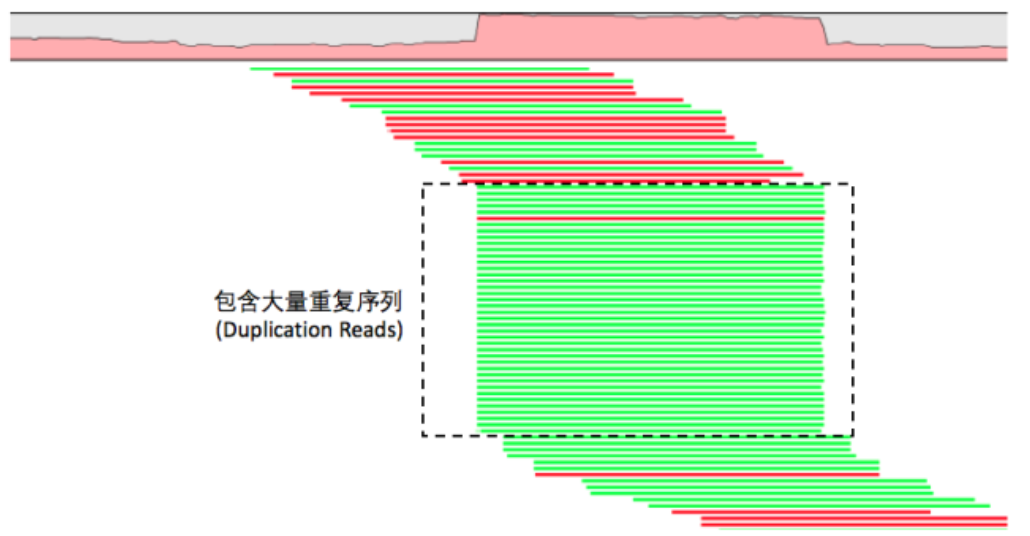
探究samtools和picard去除read duplicates的方法
1、samtools
samtools 去除 duplicates 使用 rmdup
1 | $ samtools rmdup [-sS] <input.srt.bam> <out.bam> |
只需要开始-s的标签, 就可以对单端测序进行去除PCR重复。其实对单端测序去除PCR重复很简单的,因为比对flag情况只有0,4,16,只需要它们比对到染色体的起始终止坐标一致即可,flag很容易一致。
Remove potential PCR duplicates: if multiple read pairs have identical external coordinates, only retain the pair with highest mapping quality. I
<img src=./picture/GATK4-pipeline-remove-duplicates-samtools-principle-1.png width=900/>
但是对于双端测序就有点复杂了~
In the paired-end mode, this command ONLY works with FR orientation and requires ISIZE is correctly set. It does not work for unpaired reads (e.g. two ends mapped to different chromosomes or orphan reads).
<img src=./picture/GATK4-pipeline-remove-duplicates-samtools-principle-2.png width=900/>
很明显可以看出,去除PCR重复不仅仅需要它们比对到染色体的起始终止坐标一致,尤其是flag,在双端测序里面一大堆的flag情况,所以我们的94741坐标的5个reads,一个都没有去除!
这样的话,双端测序数据,用samtools rmdup效果就很差,所以很多人建议用picard工具的MarkDuplicates 功能
2、picard
picard对于单端或者双端测序数据并没有区分参数,可以用同一个命令!
对应单端测序,picard的处理结果与samtools rmdup没有差别,不过这个java软件的缺点就是奇慢无比
操作:排序及标记重复


1、排序(SortSam)
- 对sam文件进行排序并生成bam文件,将sam文件中同一染色体对应的条目按照坐标顺序从小到大进行排序
- GATK4的排序功能是通过
picard SortSam工具实现的。虽然samtools sort工具也可以实现该功能,但是在GATK流程中还是推荐用picard实现,因为SortSam会在输出文件的头信息部分添加一个SO标签用于说明文件已经被成功排序,且这个标签是必须的,GATK需要检查这个标签以保证后续分析可以正常进行 https://software.broadinstitute.org/gatk/documentation/tooldocs/current/picard_sam_SortSam.php
1 | # 使用GATK命令 |

如何检查是否成功排序?
1 | $ samtools view -H /path/to/my.bam |
若随后的比对记录中的contig那一列的顺序与头文件的顺序一致,且在头信息中包含SO:coordinate这个标签,则说明,该文件是排序过的
2、标记重复(Markduplicates)
- 标记文库中的重复
https://software.broadinstitute.org/gatk/documentation/tooldocs/current/picard_sam_markduplicates_MarkDuplicates.php
1 | gatk MarkDuplicates -I preprocess/T.chr17.sort.bam -O preprocess/T.chr17.markdup.bam -M preprocess/T.chr17.metrics --CREATE_INDEX |

质量值校正
检测碱基质量分数中的系统错误,需要用到 GATK4 中的 BaseRecalibrator 工具
碱基质量分数重校准(Base quality score recalibration,BQSR),就是利用机器学习的方式调整原始碱基的质量分数。它分为两个步骤:
- 利用已有的snp数据库,建立相关性模型,产生重校准表( recalibration table)
- 根据这个模型对原始碱基进行调整,只会调整非已知SNP区域。
注:如果不是人类基因组,并且也缺少相应的已知SNP数据库,可以通过严格SNP筛选过程(例如结合GATK和samtools)建立一个snp数据库。
注意:Base Recalibration是以read group为单位进行的,因此当一个BAM文件中包含了多个read group时,BaseRecalibrator会对每个read group分别建立校正模型
1、建立较正模型
质量值校正,这一步需要用到variants的known-sites,所以需要先准备好已知的snp,indel的VCF文件:
1 | # 下载known-site的VCF文件,到Ensembl上下载 |
2、质量值校准
1 | # 质量校正 |
SNP、 INDEL位点识别
SNP calling 策略的选择
当你有多个samples,然后你call snp时候,你是应该将所有sample分开call完之后,再merge在一起。还是直接将所有samples,同时用作input然后call snp呢?
这里需要知道有哪些snp calling的策略:
single sample calling:每一个sample的bam file都进行单独的snp calling,然后每个sample单独snp calling结果再合成一个总的snp calling的结果。
batch calling: 一定数目群集的bamfiles 一起calling snps,然后再merge在一起
joint calling: 所有samples的BAM files一起call 出一个包含所有samples 变异信息的output
一般来说,如果条件允许(computational power等),使用joint calling ,即将所有samples同时call是比较优的选择
原因:
1、对于低频率的变异具有更高更好的检测sensitivity
在joint calling中,由于所有samples中所有的位点都是同时call的,换句话说就是,所有位点的信息都是share的,因此可以如果某些samples中个别位点是低频率的,但是可能在其他samples中,该位点的频率比较高,因此可以准确的对低频位点有更加好的calling 效果。

如左图,在1到n的samples中,碱基G只出现在其中两个samples中。如果我们将这些samples单独的call snps,这个低频率G的位点将会被忽略。但是joint calling 却可以允许将这些碱基G出现的频率进行累加,将该低频率的突变也call出来。
在右图中,上面的sample是一个跟ref一样具有纯合位点,下面的sample在一些位点中有部分数据缺少的情况,例如rs429358.如果将这两个samples分开call snp,然后merge一起,这样会错误地将这两个samples,在这个位点上看作具有类似的变异频率,但是其实由于下面的samples在改位点的区域存在数据缺少,只能看作non-informative。
2、更好的过滤掉假阳性的变异callling的结果
现在使用过滤变异的方法例如VQSR等利用的统计模型,都基于一个比较大的samples size。joint calling 这种方法可以提供足够的数据,确保过滤这一步是统一应用于所有samples的。
Germline SNPs + Indels
将一个或多个个体放在一起call snp,得到一个 joint callset,该snp calling的策略称为joint calling

第一步,单独为每个样本生成后续分析所需的中间文件——gVCF文件。这一步中包含了对原始fastq数据的质控、比对、排序、标记重复序列、BQSR和HaplotypeCaller gVCF等过程。这些过程全部都适合在单样本维度下独立完成。值得注意的是,与单样本模式不同,该模式中每个样本的gVCF应该成为这类流程的标配,在后续的步骤中我们可以通过gVCF很方便地完成群体的Joint Calling;
第二步,依据第一步完成的gVCF对这个群体进行Joint Calling,从而得到这个群体的变异结果和每个人准确的基因型(Genotype),最后使用VQSR完成变异的质控。
SNP、 INDEL位点识别
1、单独为每个样本生成后续分析所需的中间文件——gVCF文件
1 | $ gatk HaplotypeCaller -R Ref/chr17.fa -I sam/T.chr17.recal.bam -ERC GVCF --dbsnp ../Ref/VCF/dbsnp_138.hg19.vcf \ |
HaplotypeCaller可以同时call snp 和indel,通过局部 de-novo 组装而非基于mapping的结果。一旦程序在某个区域发现了变异信号,它会忽略已有的mapping信息,在该区域执行reads重组装
如何对指定的区域call snp?
使用
-L参数指定识别突变位点的区域
提供字符串指定单个interval,如
-L chr17:7400000-7800000只识别17号染色体7400000-7800000 区域的突变位点。或者使用-XL参数排除某个区域提供一个保存interval lists的文件。GATK支持多种格式的interval lists文件
Picard-style
.interval_list由 SAM-like header 和 intervals 信息组成,坐标系统为 1-based
2
3
4
5
6
7
8
9
10
> @SQ SN:1 LN:249250621 AS:GRCh37 UR:http://www.broadinstitute.org/ftp/pub/seq/references/Homo_sapiens_assembly19.fasta M5:1b22b98cdeb4a9304cb5d48026a85128 SP:Homo Sapiens
> @SQ SN:2 LN:243199373 AS:GRCh37 UR:http://www.broadinstitute.org/ftp/pub/seq/references/Homo_sapiens_assembly19.fasta M5:a0d9851da00400dec1098a9255ac712e SP:Homo Sapiens
> 1 30366 30503 + target_1
> 1 69089 70010 + target_2
> 1 367657 368599 + target_3
> 1 621094 622036 + target_4
> 1 861320 861395 + target_5
> 1 865533 865718 + target_6
>
- GATK-style `.list` or `.intervals` 这种格式很简单,intervals需要写成这种格式:`<chr>:<start>-<stop>`,坐标系统为 1-based - BED files `.bed` BED3格式:`<chr> <start> <stop>`,坐标系统为 0-based,GATK只接受 1-based 坐标系统,因此GATK会根据文件后缀 `.bed` 识别bed文件格式,然后会将 0-based 转换为 1-based **注意:** > - intervals 必须按照 reference 的坐标进行排序 > - 可以提供多个intervals集合,文件格式不必一致:`-L intervals1.interval_list -L intervals2.list -L intervals3.bed ...`
2、合并多个GVCF文件得到GenomicsDB,为joint genotyping做准备。若是单样本跳过该步骤
1 | $ gatk --java-options "-Xmx4g -Xms4g" GenomicsDBImport \ |
也可以使用CombineGVCFs实现该功能。CombineGVCFs继承自GATK4之前的版本,但是它的运行速度不如GenomicsDB
1 | $ gatk CombineGVCFs \ |
3、联合call snp,得到 joint-called SNP&indel
1 | # 单样本 |
SNP、 INDEL位点过滤
方法一:通过质量校正来过滤(Filter Variants by Variant (Quality Score) Recalibration)
1、用机器学习的方法基于已知的变异位点对caller给出的原始 variant quality score 进行校正 (VQSR),包含两步:(1)构建校正模型;(2)应用模型进行质量值校正
由于评价SNP和Indel质量高低的标准不同,因此需要分SNP和Indel两种不同的模式,分别进行校正
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48# SNP mode
## 构建校正模型
$ gatk VariantRecalibrator \
-R Homo_sapiens_assembly38.fasta \
-V input.vcf.gz \
--resource hapmap,known=false,training=true,truth=true,prior=15.0:hapmap_3.3.hg38.sites.vcf.gz \
--resource omni,known=false,training=true,truth=false,prior=12.0:1000G_omni2.5.hg38.sites.vcf.gz \
--resource 1000G,known=false,training=true,truth=false,prior=10.0:1000G_phase1.snps.high_confidence.hg38.vcf.gz \
--resource dbsnp,known=true,training=false,truth=false,prior=2.0:Homo_sapiens_assembly38.dbsnp138.vcf.gz \
-an QD -an MQ -an MQRankSum -an ReadPosRankSum -an FS -an SOR \
-mode SNP \
-O output.snps.recal \
--tranches-file output.snps.tranches \
--rscript-file output.snps.plots.R
## 应用模型进行质量值校正
$ gatk ApplyVQSR \
-R Homo_sapiens_assembly38.fasta \
-V input.vcf.gz \
-O output.snps.VQSR.vcf.gz \
--truth-sensitivity-filter-level 99.0 \
--tranches-file output.snps.tranches \
--recal-file output.snps.recal \
-mode SNP
# Indel mode
## 上一步SNP mode产生的输出作为这一步的输入
## 构建校正模型
$ gatk VariantRecalibrator \
-R Homo_sapiens_assembly38.fasta \
-V input.snps.VQSR.vcf.gz \
--resource hapmap,known=false,training=true,truth=true,prior=15.0:hapmap_3.3.hg38.sites.vcf.gz \
--resource omni,known=false,training=true,truth=false,prior=12.0:1000G_omni2.5.hg38.sites.vcf.gz \
--resource 1000G,known=false,training=true,truth=false,prior=10.0:1000G_phase1.snps.high_confidence.hg38.vcf.gz \
--resource dbsnp,known=true,training=false,truth=false,prior=2.0:Homo_sapiens_assembly38.dbsnp138.vcf.gz \
-an QD -an MQ -an MQRankSum -an ReadPosRankSum -an FS -an SOR \
-mode INDEL \
-O output.indels.recal \
--tranches-file output.indels.tranches \
--rscript-file output.indels.plots.R
## 应用模型进行质量值校正
$ gatk ApplyVQSR \
-R Homo_sapiens_assembly38.fasta \
-V input.snps.VQSR.vcf.gz \
-O output.snps.indels.VQSR.vcf.gz \
--truth-sensitivity-filter-level 99.0 \
--tranches-file output.indels.tranches \
--recal-file output.indels.recal \
-mode INDEL执行变异校正需要满足两个条件:
- 大量高质量的已知variants作为训练集,而这对于许多的物种是不满足的
- 数据集不能太小,因为它需要足够的数据集来识别 good vs. bad variants
因此对于只有一两个样本、靶向测序、RNA-seq、非模式动物的数据,都不推荐进行变异质量值校正。若以上两个条件都不满足,则需要采取直接过滤 (hard-filtering) 的策略
方法二:直接过滤 (hard-filtering)
Steps
- Extract the SNPs from the call set
- Apply the filter to the SNP call set
- Extract the Indels from the call set
- Apply the filter to the Indel call set
- Combine SNP and indel call set
- Get passed call set
提取SNP位点
1
2$ gatk SelectVariants -R Ref/chr17.fa -V calling/T.chr17.raw.snps.indels.genotype.vcf \
--select-type-to-include SNP -O filter/T.chr17.raw.snps.genotype.vcf
参数说明:
1
2
3
4
--select-type-to-include,-select-type:Type
Select only a certain type of variants from the input file This argument may be specified
0 or more times. Default value: null. Possible values: {NO_VARIATION, SNP, MNP, INDEL,
SYMBOLIC, MIXED}
- **提取INDEL位点**
1
2
$ gatk SelectVariants -R Ref/chr17.fa -V calling/T.chr17.raw.snps.indels.genotype.vcf \
--select-type-to-include INDEL -O filter/T.chr17.raw.indels.genotype.vcf
- **SNP位点过滤**
1
2
$ gatk VariantFiltration -R Ref/chr17.fa -V filter/T.chr17.raw.snps.genotype.vcf --filter-expression "QD < 2.0 || FS > 60.0 || MQ < 40.0 || SOR > 3.0 || MQRankSum < -12.5 || \
ReadPosRankSum < -8.0" --filter-name "SNP_FILTER" -O filter/T.chr17.filter.snps.genotype.vcf
参数说明:
1
2
3
4
5
6
--filter-expression,-filter:String
One or more expression used with INFO fields to filter This argument may be specified 0
or more times. Default value: null.
--filter-name:String Names to use for the list of filters This argument may be specified 0 or more times.
Default value: null.
- **INDEL位点过滤**
1
2
$ gatk VariantFiltration -R Ref/chr17.fa -V filter/T.chr17.raw.indels.genotype.vcf --filter-expression "QD < 2.0 || FS > 200.0 || SOR > 10.0 || MQRankSum < -12.5 || ReadPosRankSum < \
-20.0" --filter-name "INDEL_FILTER" -O filter/T.chr17.filter.indels.genotype.vcf
- **合并过滤后SNP、 INDEL文件**
1
2
$ gatk MergeVcfs -I filter/T.chr17.filter.snps.genotype.vcf -I \
filter/T.chr17.filter.indels.genotype.vcf -O filter/T.chr17.filter.snps.indels.genotype.vcf
- **提取PASS突变位点**
1
2
$ gatk SelectVariants -R Ref/chr17.fa -V filter/T.chr17.filter.snps.indels.genotype.vcf -O
T.chr17.pass.snps.indels.genotype.vcf -select "vc.isNotFiltered()"
参数说明:
1
2
3
--selectExpressions,-select:String
One or more criteria to use when selecting the data This argument may be specified 0 or
more times. Default value: null.
Hard-filter阈值探究
GATK4官网给出的推荐阈值:
For SNPs:
2
3
4
5
6
7
> MQ < 40.0
> FS > 60.0
> SOR > 3.0
> MQRankSum < -12.5
> ReadPosRankSum < -8.0
>
For indels:
2
3
4
5
6
> ReadPosRankSum < -20.0
> InbreedingCoeff < -0.8
> FS > 200.0
> SOR > 10.0
>
点此 查看GATK4原始网页
该阈值选择来自于GATK4官网的推荐,阈值依据于比较真 vs. 假 snp的特征值(annotation values)统计分布
One of the most helpful ways to approach hard-filtering is to visualize the distribution of annotation values for a truth set called using a particular pipeline. These distributions are sharped by both the pipeline methodology and the underlying physical properties of the sequence data; so for a given pairing of data generation technology + analysis pipeline, you can derive filtering thresholds based on what the distributions look like for the truth set
评估数据来源:1000Genomes 中的 whole genome trio
获得真/假变异的方法:
将whole genome trio用HaplotypeCaller的GVCF mode得到各样本单独的GVCF文件,然后再用GenotypeGVCFs进行joint-calling得到未过滤的VCF文件,最后用VQSR(基于统计机器学习方法)得到过滤的变异集合
将PASS的变异认为是真的,将被过滤的变异当作是假的
比较真变异与假变异的特征值(annotation values)的分布,进行评估的特征值有:QD, FS, SOR, MQ, MQRankSum 和 ReadPosRankSum
- QualByDepth (QD)
变异质量值(variant confidence (from the QUAL field))除以该位点覆盖的非同源的depth(与参考基因组不同的),即对质量值按照depth进行了标准化
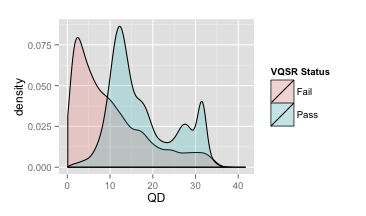
大多数被过滤掉的变异的落在低QD区域
推荐:QD≥2
- FisherStrand (FS)
一个衡量strand bias的量
1 | Phred-scaled probability that there is strand bias at the site |
当某一个位点的strand bias的程度很小或没有,那么 FS 值会接近于0

大多数过滤掉的变异的落在FS大于55的区域
推荐:FS≤60
- StrandOddsRatio (SOR)
另一种评估strand bias的量

Notice most of the variants that have an SOR value greater than 3 fail the VQSR filter
推荐:SOR≥3
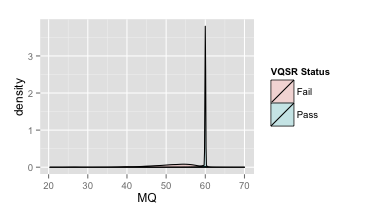
- RMSMappingQuality (MQ)
覆盖改位点的reads的mapping quality的均方根
| 原图 | 局部放大 |
|---|---|
 |
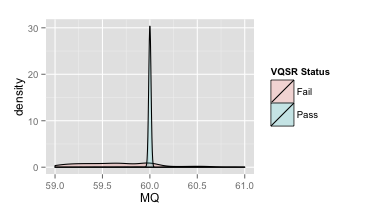 |
推荐:MQ≥40
- MappingQualityRankSumTest (MQRankSum)
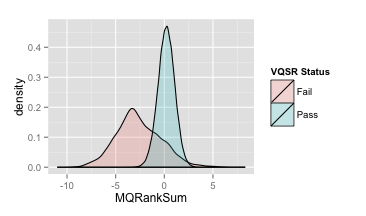
推荐:MQRankSum≥-2.5
SAMtools-BCFtools流程
关于SNP的过滤
SNP过滤的意义:
第一,过滤到一些低质量的SNP可以防止calling一些假阳性的SNP,这些假阳性的SNP会很大程度影响到后续的一系列的分析,例如GWAS等的分析,最后影响相关生物学问题的解答;
第二,如果你有很多的个体,往往你的call完SNP后,VCF文件的大小的会比较大,如果不经过过滤,对下游的群体结构分析,PCA等相关的计算,都会对计算机产生巨大的负担(运行速度还有需要的内存等)
使用vcftools进行SNP过滤
VCFtools能干什么
- 过滤特定的变异
- 比较文件
- 变异注释
- 文件格式转换
- 校验和合并文件
- 对变异位点集合进行坐标运算
参考资料:
(1) 生信菜鸟团:GATK使用注意事项
(2) 小天师兄《全外显子组测序分析》
(3) RNA-Seq是否可以替代WES完成外显子的变异检测?二代测序的四种Read重复是如何产生的?
(4) 生信菜鸟团:仔细探究samtools的rmdup是如何行使去除PCR重复reads功能的
(5) 生信技能树论坛:GATK之BaseRecalibrator
(6) GATK Forum: Collected FAQs about input files for sequence read data (BAM/CRAM)
(7) GATK Forum: What input files does the GATK accept / require?
(8) GATK Forum: Collected FAQs about interval lists
(9) GATK Forum:GATK Best Practices
(10) 公众号碱基矿工:GATK4全基因组数据分析最佳实践 ,我以这篇文章为标志,终结当前WGS系列数据分析的流程主体问题 | 完全代码
(11) 生信菜鸟团:生信笔记:call snp是应该一起call还是分开call?
(12) GATK Forum:Hard-filtering germline short variants
(13) 生信杂谈:用泊松分布解释 NGS 测序数据的 duplication 问题
(14) 【简书】关于SNP的过滤(1):如何使用vcftools进行SNP过滤
(15) VCFtools官网