Part1:lincRNA注释信息数据库
实现目标
- 将lincRNA的gtf格式的注释信息写到MySQL数据库中
- 用户需要登录才具有数据库访问权限
- 提供数据库的检索功能
思路
从 UCSC genome browser 或 Ensemble 等数据库中下载人类(homo sapiens)和小鼠 (Mus musculus)的全基因组注释信息文件(GTF或者GFF3格式文件)
提取出其中的lincRNA的注释信息记录,然后将其写入MySQL数据库系统
写好PHP脚本与之前建好的MySQL数据库系统进行数据交互
准备需要写入数据库中的数据
下载注释信息文件
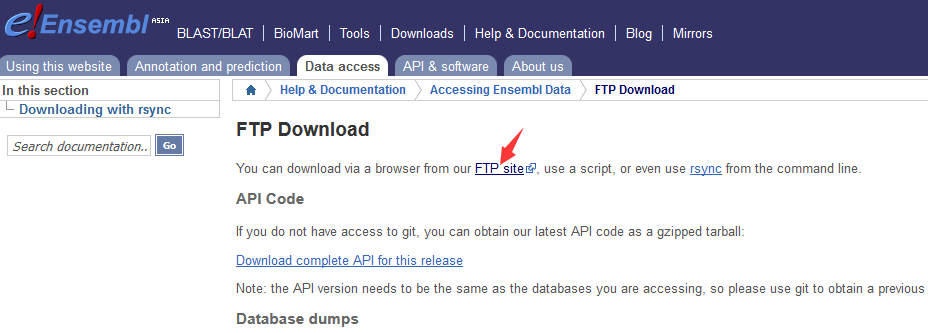
获取GTF文件的下载地址:
打开Ensemble数据库:http://asia.ensembl.org/index.html

![]()
![]()
![]()
以下列出了可供下载的基因组注释信息版本release-XX,目前的最新版本为release-91
得到下载链接,开始下载(下载至Linux服务器中)
1 | # 下载GRCh38.91(人类)注释文件 |
提取lincRNA部分记录
首先解压前面下载的压缩文件
1 | $ gunzip /Path/To/dir/Homo_sapiens.GRCh38.91.gtf.gz |
GTF格式说明
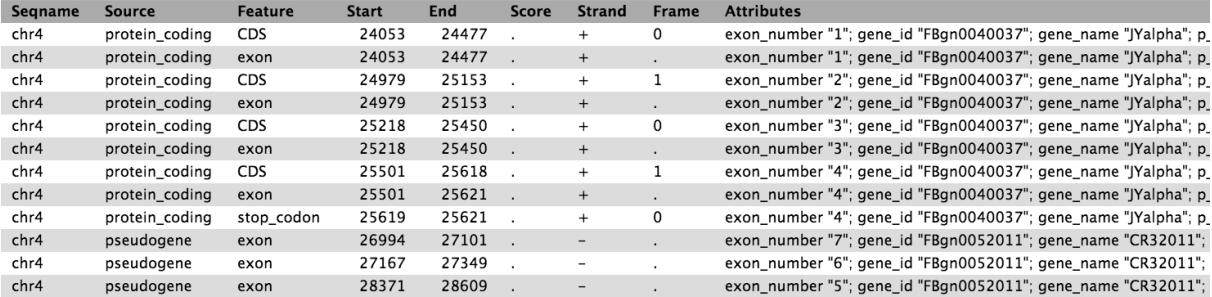
看一下我们的GTF文件的具体情况
1 | Chrom Resource Feature Start End Score Strand Frame Attributes |
可以看到,最后一列的gene_biotype中保存的是基因类型,例如protein_coding,lincRNA,miRNA等,所以针对这一列来提取
用Perl单行命令提取lincRNA注释记录
1 | $ perl -ne 'chomp;next if (/^\#/);@gtf=split /\t/;if($gtf[8] =~ /gene_biotype\s\"lincRNA\"/) {print "$_\n";}' /Path/To/dir/Mus_musculus.GRCm38.91.gtf >/Path/To/dir/lincRNA_GRCm38.91.gtf |
想获取该数据库实战中的示例数据,请点 这里
提取出来的lincRNA注释记录为以下形式:
1 | Chrom Resource Feature Start End Score Strand Frame Attributes |
接下来,将以上格式的信息格式化成以下形式:
1 | Chrom Biotype Feature Start End GeneId GeneName TranscriptId ExonNumber |
利用Perl的正则匹配提取目标字符串进行文本格式化,想了解正则表达式,请点 这里
对于第三列Feature的不同,可以分别构造不同的正则表达式:
- gene:
原格式:
2
>
正则表达式:
2
>
- transcript:
原格式:
2
>
正则表达式:
2
>
- exon:
原格式:
2
>
正则表达式:
2
>
1 | # 正则匹配的正则表达式可能有点复杂,这里为了方便阅读分为多行且加上缩进,在实际命令行中请写成一行 |
将数据写入数据库中
以下以将lincRNA_GRCh38.91.gtf.format的数据导入lincRNA_h表中为例
方法一:使用MySQLi
MySQLi:PHP中用于与MySQL数据库系统进行数据库交互的扩展
使用MySQLi的预处理与参数绑定方法,进行数据的批量导入
预备知识:
1. 首先,创建好表格
1 | # 登录MySQL |
- 编辑php脚本
lincRNA_Ano_data_batch_import.php
1 | <?php |
- 执行php脚本,完成数据的批量导入
1 | $ php -f lincRNA_Ano_data_batch_import.php <tablename> <data-to-import> |
想了解如何在 Linux 命令行中使用和执行 PHP 代码,请点 这里
方法二:使用sql脚本
准备sql脚本
要往MySQL数据库中写入数据,需要用INSERT INTO 语句
1 | INSERT INTO table_name (column1,column2,column3,...) |
一般情况下我们往数据库中写入少数几条数据时,直接在MySQL的交互环境中执行INSERT INTO 语句即可,但是当要批量导入时,再采用在MySQL的交互环境中执行INSERT INTO 语句,明显是低效而不现实的,这个时候我们可以写一个sql脚本,来执行数据库的批量操作。
简单来说sql脚本就是把多个INSERT INTO 语句写到一个文件里
sql脚本的文件起始部分(为人类和小鼠的数据分别准备一个sql文件,创建的对应表名分别为 lincRNA_h 和 lincRNA_m ,以下以人类的为例):
1 | SET NAMES utf8; |
然后,用perl单行命令实现以下转换:
1 | 1 lincRNA gene 29554 31109 ENSG00000243485 MIR1302-2HG - - |
![]()
1 | INSERT INTO lincRNA_h (chrom,biotype,feature,start,end,geneid,genename,transcriptid,exon) VALUES ("1","lincRNA","gene","29554","31109","ENSG00000243485","MIR1302-2HG","-","-"); |
并将转换后的结果追加到上一步已经写好起始部分的sql脚本的后面
1 | $ perl -ane 'chomp;print "INSERT INTO lincRNA_h (chrom,biotype,feature,start,end,geneid,genename,transcriptid,exon) VALUES (\"$F[0]\",\"$F[1]\",\"$F[2]\",\"$F[3]\",\"$F[4]\",\"$F[5]\",\"$F[6]\",\"$F[7]\",\"$F[8]\");\n"' /Path/To/lincRNA_GRCh38.91.gtf.format >>/Path/To/lincRNA_h.sql |
写入数据
登录,进入MySQL的交互环境,才能进行以下操作
1 | # 输入密码,登录MySQL |
编辑php脚本
注册
创建注册页面
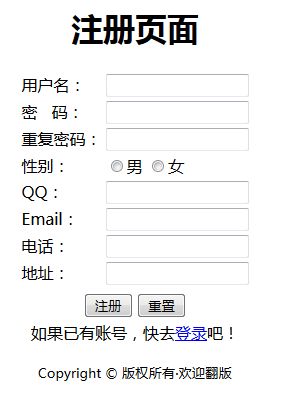
表单部分的代码(想了解表单的相关知识,请点 这里):
1 | <form action="registeraction.php" method="post"> |
表单信息提交的对象为同一目录下的registeraction.php脚本
注册信息处理脚本
registeraction.php脚本
1 | <?php |
登录
登录页面
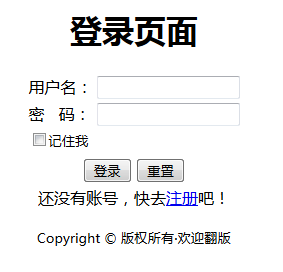
1 | <form id="loginform" action="loginaction.php" method="post"> |
登录脚本
该方法为,用root用户权限从User表中提取相应的用户名和密码信息,与用户填写的用户名和密码进行比较,来判断用户用户是否拥有数据库的访问权限
该方法需要拥有root用户的账户密码,若没有,请使用第二种方法
1 | <?php |
该方法通过使用用户填写的用户名和密码直接尝试登陆MySQL,通过登录结果来判断用户是否拥有数据库的使用权限
1 | <?php |
登录成功

通过判断全局变量$_SESSION['user']是否定义来判断是否成功登录
1 | <?php |
退出登录
1 | <?php |
检索数据库
检索数据库,需要用到数据库用户的用户名和密码,用于在之前的登录过程中,我们已经将用户名和密码存储在 $_SESSION 中,所以可以通过 $_SESSION['user'] 和 $_SESSION['password'] 获得
这里将表单部分与PHP命令写在一个PHP脚本中,脚本命名为databaseQuery_Ano.php
表单部分
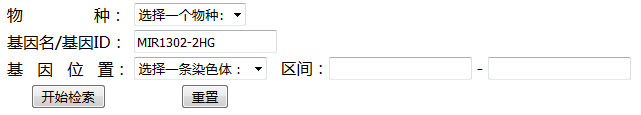
1 | <form action="#" method="post"> |
PHP脚本
1 | <?php |
Part2:内容与功能升级
实现目标
- 增加 blast 功能:将用户提交的序列与 lncRNA 的序列库进行 blast 比对
- 创建 lncRNA 表达数据库,除了 part1 中提供的基础检索功能外,还要提供绘图功能
- 增加数据库的删除、追加(单条记录追加和批量追加)与编辑功能
思路
1. 增加 blast 功能
目标序列的指定方式:
- 用户在输入框中输入
- 用户提交目标序列的 fasta 文件
创建lncRNA的 blast 序列库(即lncRNA的 blast 索引):
- 根据 part1 《准备需要写入数据库中的数据》 中得到的文件
lincRNA_GRCm38.91.gtf.format和lincRNA_GRCh38.91.gtf.format,从中提取unique的lincRNA的Ensembl gene id - 利用lincRNA的Ensembl gene id,从Ensembl的BioMart中下载lincRNA对应的序列
- 对这些序列建立blast索引
2. 创建 lncRNA 表达数据库
从公共数据库(如GEO)中直接下载RNA-seq表达谱文件,或者下载原始fastq文件,自己跑RNA-seq分析流程来获取表达谱,然后提取其中的lincRNA部分,进行差异表达分析后将差异表达结果写进MySQL数据库中,同时保留表达谱文件,以供后期绘图时访问
表达数据来源:从GEO数据库中下载某个RNA-seq实验的表达谱文件
差异表达分析:使用DESeq2进行差异表达分析,DESeq2使用方法请点 这里
将差异表达结果写进MySQL数据库中:过程与 part1 《将数据写入数据库中——方法一:使用MySQLi》一样
绘图:即在检索获得差异表达结果列表后,每条记录后提供一个绘图按钮,用户点击后即可绘制出该基因在不同samples的表达柱状图,可通过调用R脚本实现
3. 增加数据库的删除、追加与编辑功能
这部分应该是整个part2中最容易实现的部分
删除 —— 注意:在真正删除之前,请要求用户进行再次确认
- 删除一条记录:用SQL的DELETE语句
- 删除多条记录:在每条记录前有一个复选框,选中多条记录前的复选框,然后点击页面某个角落的“Delete”按钮,提交删除操作请求
追加
- 追加一条记录
- 批量追加:可以让用户提交文件来实现批量追加,不过真正进行追加操作之前,务必进行文件格式的检查
编辑 —— 用UPDATE语句
添加blast功能
获得chrX上lincRNA序列
- 利用Gene stable ID批量下载序列
第一次写作业时,在mysql数据库中添加了lncRNA的位置信息、ID等。这次从之前建立数据得到的lincRNA_GRCh38.91.gtf.format和lincRNA_GRCm38.91.gtf.format文件中提取Gene stable ID,批量下载FASTA格式的序列信息。
- 得到lnRNA的ID
先以lincRNA_GRCh38.91.gtf.format(人类)为例
1. 通过less lincRNA_GRCh38.91.gtf.format查看文件
实例如下,第6列的就是基因ID(ENSG00000243485开始)
1 | 1 lincRNA gene 29554 31109 ENSG00000243485 MIR1302-2HG - - |
2. 用cut -f 6 lincRNA_GRCh38.91.gtf.format|sort|uniq > lincRNA_GRCh38.91.geneid.txt命令截取第6列ID信息,并输出到out文件中
3. 打开ensembl的biomart,按照图片调整参数,最后上传ID信息文件即可。
(由于全基因组数据太大,所以我在REGION中只选择了x染色体)
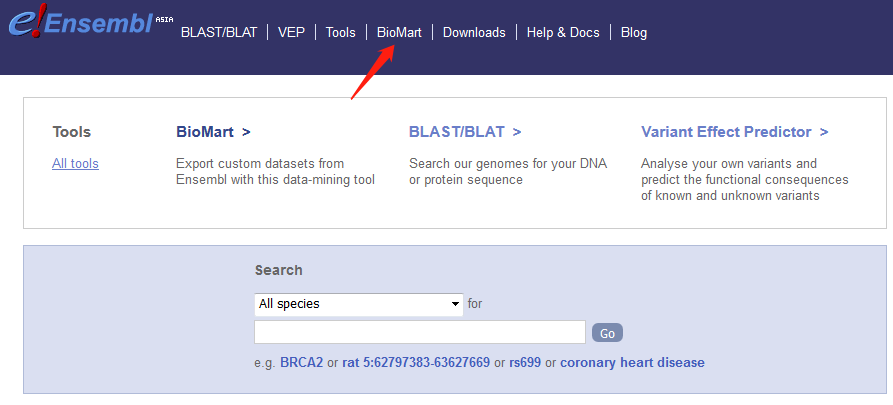
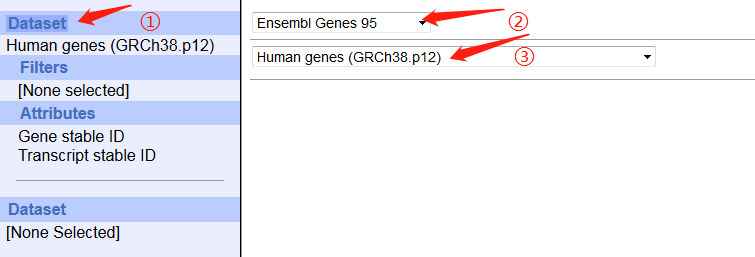
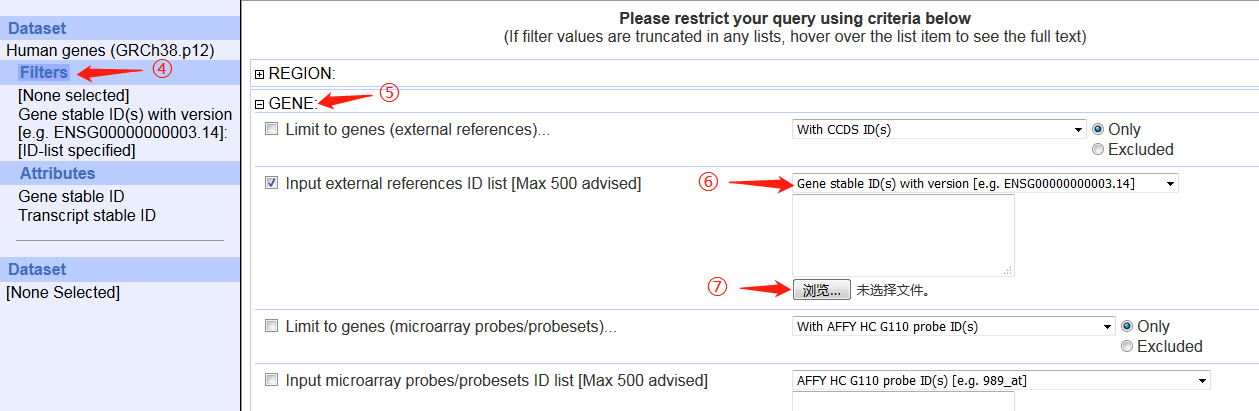
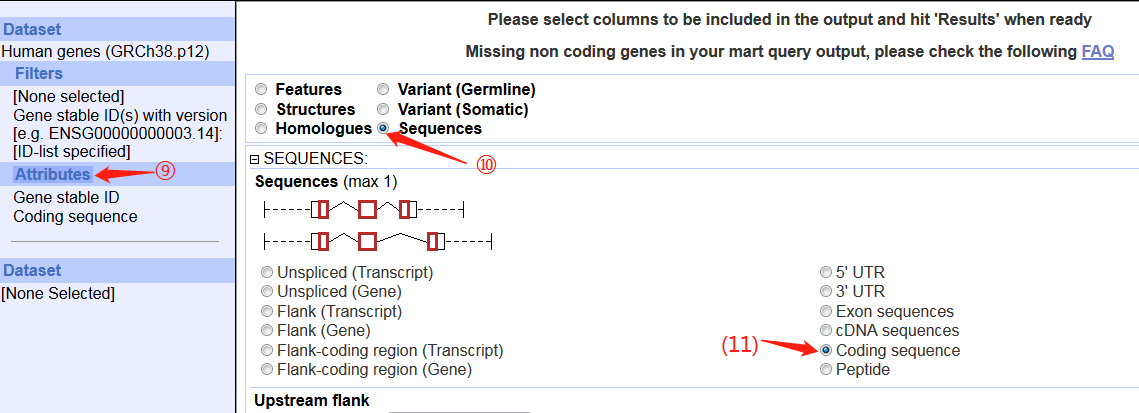
4. 点击results,跳转页面点GO生成FASTA序列。
5. 小鼠数据如法炮制,只是在第一张照片处把Human参数改成Mouse。
构建BLAST索引
- 删除课堂教学用的数据
每人配额只有2G,所以清空一下磁盘,方便做作业
1 | #查看文件大小 |
- 构建BLAST索引
在biomart下载完FASTA格式序列后,会得到mart_export.txt文件,修改文件名为lincRNA_chrX.fa将其导入服务器。
调用BLAST命令构建索引
1 | makeblastdb -in /Path/To/lincRNA_chrX.fa -input_type fasta -dbtype nucl -title lncRNA_h -parse_seqids -out /Path/To/lncRNA_h -logfile File_h |
参数介绍
- -in:FASTA文件位置
- -input_type:输入格式
- -dbtype:数据库格式
- -title:索引名称
- -parse_seqids:不知道啥意思
- -out:输出文件名(title感觉没啥用,最后blast调用索引都是调用输出文件)
- -logfile:软件日志
命令完成,打开软件日志File_h查看:
1 | Building a new DB, current time: 05/26/2018 15:17:13 |
成功!
执行BLAST
- 提交BLAST检索申请,脚本保存为
blastQuery.php
1 | <form action="localblast.php" method="post" enctype="multipart/form-data"> |
- 执行BLAST操作,保存为
localhost.php
1 | <?php |
添加lincRNA表达谱
获取在两个样本组的表达谱
数据集简单介绍与下载
该笔记中使用的数据集为 Nature Protocols 文章中用到的数据集
| sample id | Sex | population |
|---|---|---|
| ERR188245 | Female | GBR |
| ERR188428 | Female | GBR |
| ERR188337 | Female | GBR |
| ERR188401 | Male | GBR |
| ERR188257 | Male | GBR |
| ERR188383 | Male | GBR |
| ERR204916 | Female | YRI |
| ERR188234 | Female | YRI |
| ERR188273 | Female | YRI |
| ERR188454 | Male | YRI |
| ERR188104 | Male | YRI |
| ERR188044 | Male | YRI |
文章作者已经将数据集打包好了,下载链接:ftp://ftp.ccb.jhu.edu/pub/RNAseq_protocol/chrX_data.tar.gz
1 | # 下载 |
解压后可以看到数据集的组成1
2
3
4
5
6
7
8
9|---chrX_data
|---genes
|---chrX.gtf
|---genome
|---chrX.fa
|---indexes
|---chrX_tran.[1-7].ht2
|---samples
|---ERR*_chrX_[12].fastq.gz
跑RNA-seq分析流程
RNA-seq分析流程,请参考 这里
一般第一步是创建hisat2索引,由于数据集中已经提供了(在chrX_data/indexes文件夹下),所以跳过这一步
- hisat2比对
在比对前,先准备好一个保存样本Id的文本samplelist.txt:
1 | ERR188245 |
开始执行批量比对
1 | $ mkdir map |
- stringtie转录本拼接
1 | $ mkdir asm |
- stringtie定量
以read count进行定量,作为DESeq2或edgeR的输入
1 | $ mkdir quant |
最后会在工作目录下产生两个read count定量文件:
- gene定量:gene_count_matrix.csv
- transcript定量:transcript_count_matrix.csv
想下载这两个文件,请点 这里
- 差异表达分析:DESeq2
DESeq2要求输入的表达矩阵是read counts
构建 DESeqDataSet 对象
1 | # 构建表达矩阵 |
差异表达分析
1 | dds <- DESeq(dds) |
这一步最终得到差异表达分析结果文件gene_diffResult.txt,想下载这两个文件,请点 这里
将差异表达结果写进MySQL
过程与 part1 方法一:使用MySQLi 相同
1 | # 创建表格DiffExp_result |
| 数据类型 | 描述 |
|---|---|
| TINYINT(size) | -128 到 127 常规。0 到 255 无符号*。在括号中规定最大位数。 |
| SMALLINT(size) | -32768 到 32767 常规。0 到 65535 无符号*。在括号中规定最大位数。 |
| MEDIUMINT(size) | -8388608 到 8388607 普通。0 to 16777215 无符号*。在括号中规定最大位数。 |
| INT(size) | -2147483648 到 2147483647 常规。0 到 4294967295 无符号*。在括号中规定最大位数。 |
| BIGINT(size) | -9223372036854775808 到 9223372036854775807 常规。0 到 18446744073709551615 无符号*。在括号中规定最大位数。 |
| FLOAT(size,d) | 带有浮动小数点的小数字。在括号中规定最大位数。在 d 参数中规定小数点右侧的最大位数。 |
| DOUBLE(size,d) | 带有浮动小数点的大数字。在括号中规定最大位数。在 d 参数中规定小数点右侧的最大位数。 |
| DECIMAL(size,d) | 作为字符串存储的 DOUBLE 类型,允许固定的小数点。 |
基于 part1 中的php脚本data_batch_import.php稍作修改即可复用
需要改动的位置如下图:
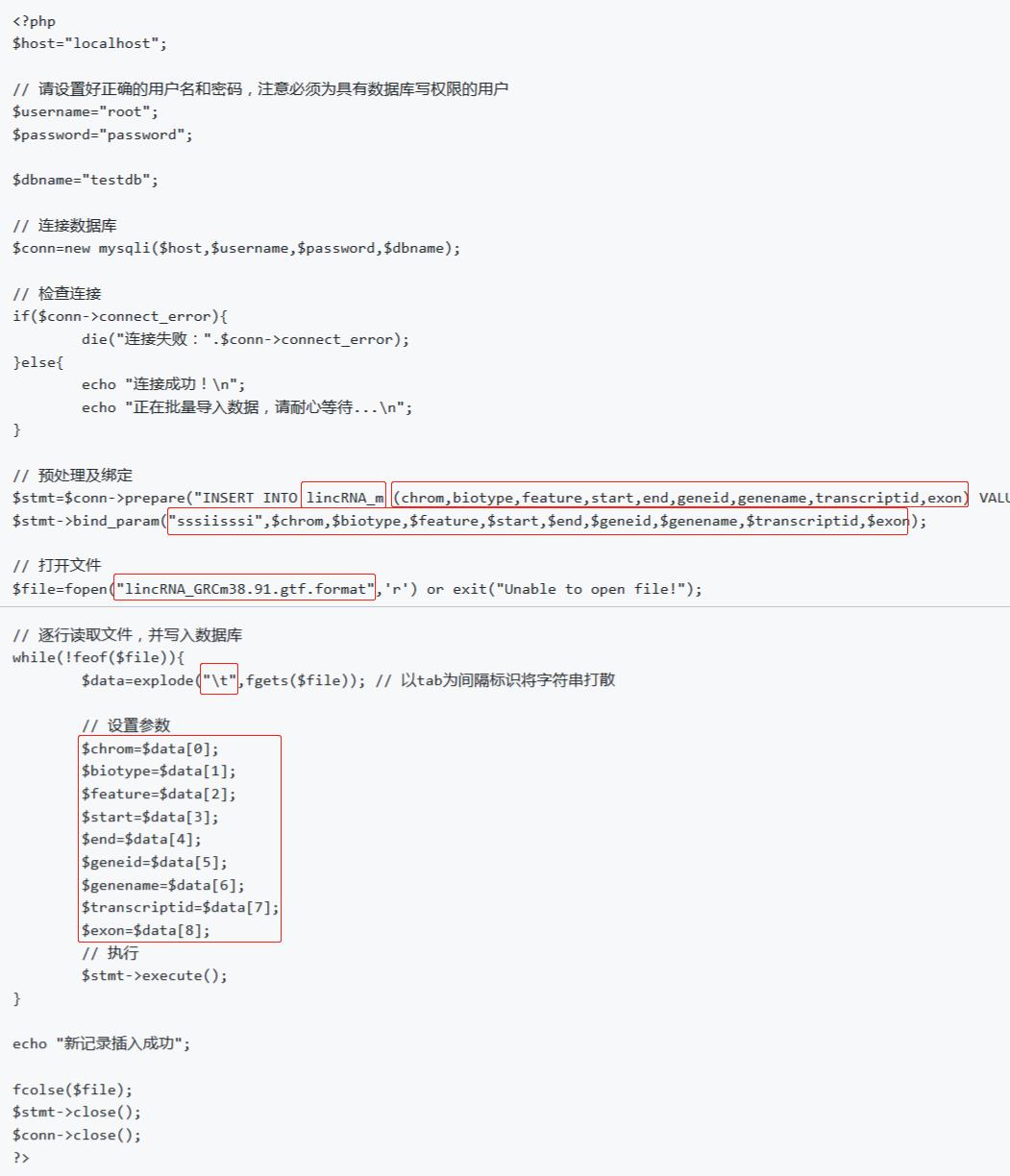
直接手动修改PHP脚本,保存为lincRNA_DiffExp_data_batch_import.php
以下只给出修改部分的代码
1 | // 预处理及绑定 |
最后执行php脚本,完成数据的批量导入
1 | $ php -f lincRNA_DiffExp_data_batch_import.php <data-to-import> |
检索表达谱数据库
过程类似于 part1 检索数据库
表单部分,提供以下检索筛选条件
- log2FoldChange 一般认为其绝对值大于1,差异显著
- p-value 一般认为其值小于等于0.05,差异显著
- q-value 一般认为其值小于等于0.05,差异显著
1 | <form action="#" method="post"> |
php脚本部分
1 | <?php |
画图:单个基因表达谱
实现思路:
- 用户在检索页面的记录栏的最后一项点击
Barplot以后,向绘图脚本(barplot.php)传递对应记录的RefSeq绘图脚本(
barplot.php)根据传递过来的RefSeq,调用绘图用的底层R脚本(barplot.R),调用的同时传递参数——RefSeq值
barplot.R实现的功能:读取表达谱文件(跑RNA-seq分析流程:stringtie定量 产生的gene_count_matrix.csv文件),根据传递过来的RefSeq值匹配出对应的基因的表达谱,然后用ggplot把图画出来,并将图保存为图像文件- 绘图脚本(
barplot.php)将图片用<img>标签呈现给用户
- 绘图脚本(
barplot.php)
1 | <?php |
- 底层R脚本(
barplot.R)
注意:
该脚本底层依赖
ggplot2包,若未安装请提前安装好,推荐以下两种安装方法:
- 直接在R的交互环境下,使用
install.packages("ggplot2")- 使用conda进行安装
1 | library(ggplot2) |
添加批量提交数据功能
基本的实现思路为:
提供文件提交的输入框,同时让用户指定向哪个数据库(实际上是不同的表格)提交数据。将用户提交的文件保存为服务器某个目录下的临时文件,然后用
- part1 将数据写入数据库中:方法一:使用MySQLi的脚本
lincRNA_Ano_data_batch_import.php- 或者,part2 将差异表达结果写进MySQL的脚本
lincRNA_DiffExp_data_batch_import.php将临时文件的数据追加导入相应的表格中
预备知识:
- 文件上传,请点 这里
要实现文件上传,首先要在php脚本所在的文件夹下创建一个权限全开的upload文件夹
1 | $ mkdir upload |
接着开始编辑php脚本,保存为batch_submit_data.php
表单部分
1 | <form action="batch_submit_data.php" method="post" enctype="multipart/form-data"> |
php脚本
1 | <?php |
添加Update功能
实现思路:
- 用户在检索页面的记录栏的最后一项点击
update以后,跳转到update表单填写页面(update_form.php脚本),同时向该php脚本传递对应记录的id- update表单填写页面(
update_form.php脚本)根据传递过来的id,把对应记录的信息以表单形式呈现,同时表单的每个输入框可以收集用户的修改信息- 用户修改后点击”submit”,执行update操作(
updata_operate_Ano.php或updata_operate_DiffExp.php脚本),同时将操作状态告知用户:是不是成功了?
- 脚本
update_form.php
1 | <?php |
- 脚本
update_operate_Ano.php
1 | <?php |
- 脚本
updata_operate_DiffExp.php
1 | <?php |
番外篇:前端优化——html+css
登录界面优化
实现效果:
山寨主流网站的登录界面,即将页面分为左右两半,左侧放置图片,右侧放置登录信息输入框
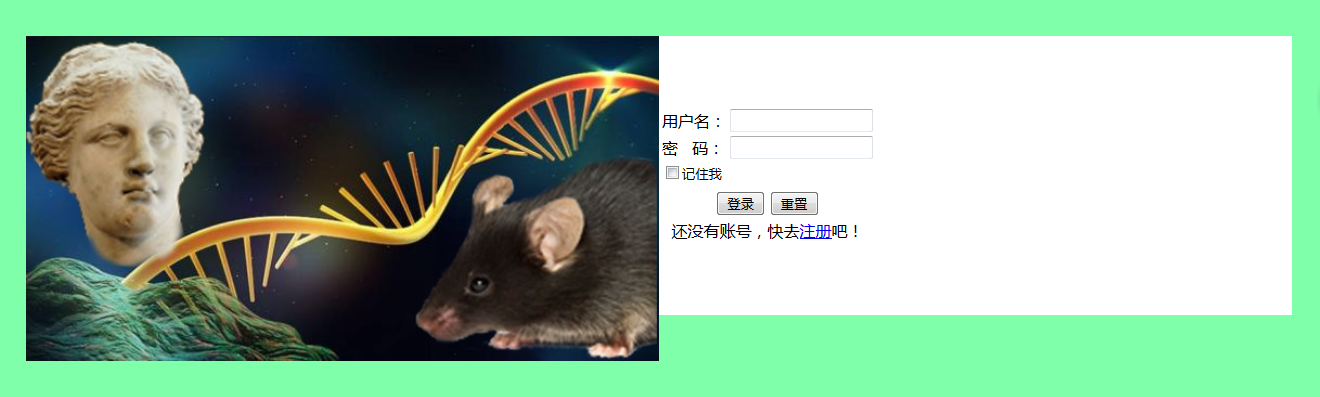
1 | <head> |
导航条菜单的制作
实现效果:
将页面分为左右部分,左侧放置垂直导航栏,右侧放置主面板
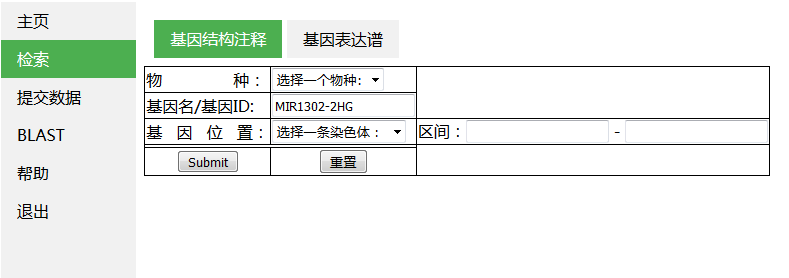
使用外部css制定页面渲染风格,css文件保存为private.css
1 | /* 导航栏样式 */ |
则在每个php脚本前加入以下代码:
1 | <head> |
参考资料: